

概要
ウェスタンブロット法は、SDS-PAGE 法でタンパク質を分子量の違いによって分離した後、PVDF メンブレンなどにタンパク質を転写し、検出したいタンパク質の特異的抗体を用いて、タンパク質を可視化する方法である。研究室でよく用いられているウェスタンブロット法は、1970年に Laemmli によって報告された条件(1)で SDS-PAGE を行った後に、転写を行うことが多い。しかし、分子量が 15 kDa 以下のタンパク質は検出しづらくなるといったことが起こる。Okajima らは分離ゲルのトリス濃度や SDS-PAGE における泳動バッファーのトリス濃度とグリシン濃度について検討し、低分子量タンパク質のバンドがクリアになることを CBB 染色で報告している(2)。この方法で SDS-PAGE を行った後、ウェスタンブロット法を行うことで低分子量タンパク質のバンドがクリアになり、上述の問題点が改善される。ここでは、グリシンとトリスを使用した Okajima らの方法に基づいたウェスタンブロット法について記述する。
イントロダクション
ウェスタンブロット法(別名イムノブロット法)は、約40年前に報告された方法であるが、現在においても生化学分野や分子生物学分野をはじめとした実験系の研究室において最も頻繁に使用されているタンパク質の解析方法の一つである(3)。
ウェスタンブロット法は、SDS-PAGE でタンパク質を分子量の違いによって分離した後、PVDF メンブレンなどにタンパク質を移し、検出するタンパク質の特異的抗体を用いて、タンパク質を可視化する方法である。しかし、ウェスタンブロット法において、おおよそ 15 kDa 以下の低分子量タンパク質は、15 kDa 以上のタンパク質と比較して、検出しづらくなるといったことが起こる。補足すると低分子量タンパク質だけではなく 100 kDa 以上の高分子量タンパク質でも検出しづらくなるといったことが起こるが、高分子量タンパク質の検出方法は別の論文や著書を参考にしていただきたい。転写バッファー中のメタノール濃度を高めると転写効率が上がる傾向にあり、低分子量タンパク質を検出しやすくなる。通常は、PVDF メンブレンでは転写バッファー中のメタノール濃度を15%以下、ニトロセルロースメンブレンでは転写バッファー中のメタノール濃度を20%以下にして転写を行うことが多い。また、高分子量タンパク質では、転写効率が下がるため、ゲルからタンパク質を溶出させやすくするために転写バッファー中に SDS を0.1%以下の濃度で加えることがあるが、低分子量タンパク質では SDS を加えないほうがメンブレンへの吸着力が良い。さらに、メンブレンの種類別に比較すると、ニトロセルロースメンブレンはバックグラウンドが抑えられ、きれいなバンドが得られやすいが、タンパク質の結合能力が弱く、低分子量タンパク質は検出しにくい。従って、低分子量タンパク質を検出する際は、ニトロセルロースメンブレンよりもタンパク質の結合能力が高い PVDF メンブレンを選択したほうが良い。また、PVDF メンブレンとニトロセルロースメンブレンともに、孔径が異なったものが販売されており、孔径が 0.45 μm、0.2 μm、0.1 μm などのメンブレンがある。低分子量タンパク質は、孔径が小さいほうが、転写においてメンブレンに吸着しやすく、メンブレンをすり抜けにくくなるので、低分子量タンパク質を解析するためには、孔径の小さいメンブレンを選ぶと良い。ただし、孔径の小さいメンブレンはバックグラウンドが上がりやすいことを念頭に置く必要がある。
低分子量タンパク質を解析するための SDS-PAGE は、トリシンを使用する方法もあるが、グリシンと比較してトリシンは高価であることや、グリシンを使用した SDS-PAGE と比較してトリシンを使用した SDS-PAGE は解析に時間を要する。Okajima ら(2)は分離ゲルのトリス濃度や SDS-PAGE における泳動バッファーのトリス濃度とグリシン濃度について検討し、Laemmli によって報告された条件(1)と比較して低分子量タンパク質のバンドがクリアになることを CBB 染色で報告している。これまで、上記に記載した転写バッファー中の試薬濃度の調節や、使用するメンブレン自体を検討することによって、ウェスタンブロット法における低分子量タンパク質の検出効率を改善できることが知られていたが、筆者は、Okajima らの方法に基づいて SDS-PAGE を行い、ウェスタンブロット法で解析したところ低分子量タンパク質の検出効率が改善された。すなわち、この結果は、SDS-PAGE における分離ゲル中のトリス濃度や泳動バッファー中のトリス濃度やグリシン濃度を調節することによっても低分子量タンパク質の検出効率が改善することができることを示すものである。本論文では、低分子量タンパク質を解析するための、トリシンを使用しない、グリシンとトリスを使用した Okajima らに基づく SDS-PAGE を用いたウェスタンブロット法について記述する。
実験器具・装置
- ゲル板(泳動用ガラスプレート MAB-10 と MB-00)(アトー)
- シールガスケット(RMS-01)(アトー)
- ラピダスマグネクリップミニ(製品番号:2398239)(アトー)2個。ゲル板が固定できれば市販の大きめのダブルクリップでもよい。市販のダブルクリップを使用する場合は4個必要。
- コーム(RM 10-12)(アトー)
- 電気泳動装置(泳動槽)(AE-6500)(アトー)
- パワーサプライ(各社から販売されているもの)
- リード線
- 50 µl もしくは 25 µl のハミルトンマイクロシリンジ(筆者は25 µl のハミルトンマイクロシリンジ 702SNR<80465>を使用)
- スパーテル
- ピンセット
- ディッシュ(プラスチック製)
- 転写準備のための容器(横 18 cm、縦 26.5 cm、高さ 5.5 cm)
- 転写装置(BE-351W)(バイオクラフト)
- 振とう機
- 発光撮影装置
試薬
- H2O(純水)
- 30%アクリルアミドミックス(29%アクリルアミド、1% N,N’-メチレンビス(アクリルアミド))
- 3.0 M トリス-HCl(pH 8.8)
- 1.0 M トリス-HCl(pH 6.8)
- 10% SDS(Sodium dodecyl sulfate)
- 10% APS(Ammonium persulfate)(小分けにして凍結保存しておくと便利)
- TEMED (N,N,N’,N’-テトラメチルエチレンジアミン)
- エタノール
- 4×SDS-PAGE サンプルバッファー(400 mM トリス HCl(pH 6.8)、8% SDS、40% グリセロール、24% 2-メルカプトエタノール、0.1% ブロモフェノールブルー)
- 泳動バッファー(50 mM トリス、380 mM グリシン、0.1% SDS)(2×泳動バッファーを作製しておくと便利)
- メタノール
- パラフィルム
- 転写バッファー(20 mM トリス、150 mM グリシン)(10×転写バッファーを作製しておくと便利)
- 転写用ろ紙(CB-09A 85×90 mm)(アトー)
- PVDF メンブレン(イモビロン-P、0.45 µm、IPVH00010)(ミリポア)、あらかじめ分離ゲルと同じぐらいの大きさに切っておく。
- TBST(140 mM 塩化ナトリウム、25 mM トリス-HCl(pH 7.4)、0.05% Tween20)または、PBST(140 mM 塩化ナトリウム、8.1 mM リン酸水素二ナトリウム、2.7 mM 塩化カリウム、0.05% Tween20)
- スキムミルク、BSA などのブロッキング剤または市販のブロッキング剤
- 1次抗体
- 2次抗体(HRP 標識)
- 化学発光検出試薬
実験手順
- ゲルの作成
- サンプルの作製
- SDS-PAGE
- ウェスタンブロット法
実験の詳細
1. ゲルの作成
Okajima らの方法(2)によるゲル作製のために必要な試薬の量を、表1に示した。低分子量タンパク質を分離するためには、15%程度かそれ以上のアクリルアミドゲルで解析することになるが、参考までに6%–12%のゲルの組成についても表1に記載した。また、表2には Laemmli 法(1)、Okajima らの方法(2)における試薬の濃度を記載した。Laemmli 法の分離ゲルと比べて、Okajima らの方法の分離ゲルはトリス-HCl(pH 8.8)の濃度が2倍である。以下に手順を記載する。
ゲル板の汚れを取り除くために、ゲル板の内側に70%エタノールを吹き付けて、キムワイプで拭く。ゲル板(MAB-10)にシールガスケットをはめこみ、ゲル板(MB-00)を重ねて、ラピダスマグネクリップミニでゲル板を固定する。
表1の分離ゲルの組成に従い、H2O、30%アクリルアミドミックス、3.0 M トリス-HCl(pH 8.8)、10% SDS、10% APS、TEMED の順に15 ml チューブに加え、転倒混和する。調製した分離ゲル溶液 5.5 ml(アトーの泳動用ガラスプレート MAB-10 と MB-00 の場合)をゲル板に流し込む。そして、分離ゲルの上にエタノールを重層し、分離ゲルが固まるまで静置する(30分から60分間)。分離ゲルが固まったら、濃縮ゲルを作製するために、表1の濃縮ゲルの組成に従い、H2O、30%アクリルアミドミックス、1.0 M トリス-HCl(pH 6.8)、10% SDS、10% APS、TEMED の順に 15 ml チューブに加え、転倒混和する。ゲル板を逆さまにして、分離ゲルの上に重層したエタノールを捨てる。調製した濃縮ゲル溶液をゲル板の一番上まで流し込み、コームを差し込み、濃縮ゲルが固まるまで静置する(30分から60分間)。ゲルは1日程度なら、ラップで包んで冷蔵保存可能である。
2. サンプルの調製
組織や培養細胞からタンパク質を抽出したサンプルあるいは精製したタンパク質などサンプルのタンパク濃度を測定し、解析するタンパク質量を見積もる。サンプルに適宜 H2O を加え、液量を調節し、サンプルと4×サンプルバッファーを 3 : 1 の割合で混合し(1.5 ml チューブを使用)、キャップロックでチューブのふたを留め、ヒートブロックを用いて95 ℃で5分間加熱する。この加熱によりタンパク質の高次構造が壊れる。加熱後すぐにキャップロックを取り外すと、加熱によって蒸発した水によって、チューブ内部の圧力の上昇により、1.5ml チューブのふたが開いてしまうことがあるため、サンプルが少し冷えるまで待つ。加熱後は短時間の遠心(フラッシュ)によってサンプル溶液全量をチューブの下側に集める。
3. SDS-PAGE
電気泳動装置の下側に泳動バッファーを加える。1. で作成したゲルのゲル板からクリップ、シールガスケット、コームをはずし、ゲル板を電気泳動装置にセットする。ゲル板の下側に空気が入っていたら電気泳動装置を斜めにして空気を抜く。電気泳動装置の上側に泳動バッファーを加える。ハミルトンシリンジの針がゲルに突き刺さらないようウェル内に入れ、ハミルトンシリンジで泳動バッファーを吸って勢いよく出す操作を何度も繰り返し、ウェル内の未重合のアクリルアミドやゲルの破片を取り除く。ハミルトンシリンジを用いて各サンプルを別々のウェルに入れていく。また、分子量マーカーを入れるウェルを残しておき、そのウェルに分子量マーカーを加える。電気泳動装置とパワーサプライをリード線でつなぎ、20 mA(定電流)で泳動する。BPB がゲルの先端近くまで移動したら、電源を切る。なお、SDS-PAGE を行っている間に、PVDF メンブレンの親水処理を行っておく(下記のウェスタンブロット法の手順に記載)。
4. ウェスタンブロット法
SDS-PAGE を行っている間に、10×転写バッファーを希釈し、1×転写バッファー 1 ℓ の調製と PVDF メンブレンの浸水処理を行う。ピンセットで、PVDF メンブレンの右端上の角を掴んで、ディッシュに移す(左利きの人は左端上の角でよい)。油性ダーマトグラフで右端上の角に印をつける(表裏を区別するためである)。以後できるだけ印をつけた部分をピンセットで掴むようにする。PVDF メンブレンに、メタノールが浸るまで加えて、よく馴染ませて、1分間静置する。メタノールを除き、転写バッファーを PVDF メンブレンが浸るまで加える。PVDF メンブレンが転写バッファーをはじき、浮き上がってくることがあるので、PVDF メンブレンと同じぐらいの大きさに切ったパラフィルムをかぶせて、PVDF メンブレンを転写バッファーに馴染ませ、SDS-PAGE が終わるまで静置しておく。PVDF メンブレンは以後乾かさないようにする。
SDS-PAGE が終了したら、転写の準備に移る。容器(横 18 cm、縦 26.5 cm、高さ 5.5 cm)にディッシュに加えなかった残りの転写バッファーを全量移し、パット(スポンジ)とろ紙を転写バッファーに入れ、手袋をはめた手で押し空気を抜く。マイナス(-)極側からパット、ろ紙(2枚)、分離ゲル、PVDF メンブレン、ろ紙(2枚)、パットの順にゲルホルダーに載せていき(図1)、すべて載せたらゲルホルダーで挟む。このときに、分離ゲルと PVDF メンブレンの間に空気が入っているとその部分の転写がうまくいかないので、空気が入っている場合は、プラス(+)極側のろ紙とパットをかぶせる前に、PVDF メンブレンと分離ゲルを手袋をはめた手で押して空気を除く。なお、SDS-PAGE 終了後のゲルの切り方については図2に示したので参考にしていただきたい。ゲルホルダーの+と-の向きを間違えないように転写装置に移し、容器とディッシュの転写バッファー全量を転写装置に加えた後、転写装置とパワーサプライをリード線でつなぎ、100 V の定電圧で1~2時間通電する。
転写している間に、ブロッキングのための5%スキムミルク溶液または5% BSA 溶液を TBST か PBST で 25 ml 調製しておく。なお、リン酸化タンパク質を検出する場合は、BSA を TBST で希釈したものをブロッキング剤として使用する(工夫とコツを参照)。また、市販のブロッキング剤を使用することで、検出時のバックグラウンドが軽減されることもある。転写が終了したら、ゲルホルダーを転写装置から取り出した後、ピンセットで PVDF メンブレンをディッシュに移し、ブロッキング溶液を加えて、振とう機を用いて60分間以上室温で振とうし、ブロッキングする(冷蔵庫で一晩反応させることも可能である)。ブロッキング終了後、ブロッキング剤を捨て、TBST もしくは PBST を PVDF の入っているディッシュに加えて、振とう機で5分間振とうして洗浄する。洗浄液を捨て、同様の操作でさらに2回以上洗浄する。
ピンセットで PVDF メンブレンを乾いたディッシュに移し、TBST または PBST で希釈した1次抗体(上記に記載したように、リン酸化タンパク質を検出する抗体の場合は TBST で希釈する)をピペットで PVDF メンブレンの上に垂らし、ディッシュを少し傾けるなどしてよく馴染ませる。抗体の使用量を節約する場合は少量の抗体希釈液を PVDF メンブレンに垂らし、よく馴染ませて、ディッシュを傾けて抗体溶液をディッシュの端に集めて、ピペットで吸って、その溶液を再び PVDF メンブレンに垂らし、よく馴染ませるといった操作を繰り返す。その後、PVDF メンブレンの上に PVDF メンブレンより少し大きめのパラフィルムを載せ、空気を抜きぴったりと密着させる。室温で1時間から2時間程度静置する(冷蔵庫で一晩反応も可能)。抗体反応終了後、TBST もしくは PBST をディッシュに加えて、振とう機で5分間振とうして PVDF メンブレンを洗浄する。洗浄溶液を捨て、同様の操作でさらに2回以上洗浄する。
ピンセットで PVDF メンブレンを乾いたディッシュに移し、TBST または PBST で希釈した2次抗体を PVDF メンブレンの上に垂らし、ディッシュを少し傾かせるなどしてよく馴染ませる。抗体の使用量を節約する場合は一時抗体の時と同様の操作を行うと良い。その後、PVDF メンブレンの上に PVDF メンブレンより少し大きめのパラフィルムを載せ、空気を抜きぴったりと密着させる。室温で1時間静置する。2次抗体の反応時間が長くなると検出時にバックグラウンド上がるためタイマーで時間を計っておいたほうが良い。抗体反応終了後、TBST もしくは PBST をディッシュに加えて、振とう機で5分間振とうして PVDF メンブレンを洗浄する。洗浄溶液を捨て、同様の操作でさらに2回以上洗浄する。
洗浄終了後、PVDF メンブレンの洗浄溶液をよく切る。PVDF メンブレンの上にピペットを用いて発光試薬(筆者は Wako のイムノスター LD を使用)を垂らし、ピンセットで PVDF メンブレンを持ち上げ傾かせるなどしてよく馴染ませる。その後、発光撮影装置で、タンパク質を検出する。
実験例
脂質ラフトの一種であるカベオラに局在するタンパク質である caveolin-1 の N 末端側より102番目から134番目のアミノ酸配列(Cav1102-134)を導入した pCMV-HA ベクター(Cav1102-134 in pCMV-HA ベクター)(4)を子宮頸癌細胞である HeLa 細胞にトランスフェクションし、48時間培養した。細胞を回収し、UltraRIPA kit for Lipid Raft(BioDynamics Laboratory Inc.)を用いて、脂質ラフトタンパク質を抽出し、上記の方法でウェスタンブロット法を行った。
転写の条件を揃えるため、SDS-PAGE 後にタンパク質をアプライしていない余分なレーンはゲルを切って除き、Okajima らの方法で SDS-PAGE を行ったゲルに含まれるタンパク質と Laemmli 法によって SDS-PAGE を行ったゲルに含まれるタンパク質を同じ PVDF メンブレンに転写した。ブロッキング剤は、PVDF Blocking Reagent for Can Get Signal(TOYOBO)を使用しブロッキングを行った。1次抗体は抗 HA マウスモノクローナル抗体(10,000倍希釈)(SIGMA-ALDRICH H9658)、2次抗体は HRP 標識された抗マウス IgG (IBL #17601)を使用し、化学発光法で HA-Cav1102-134(約 5.9 kDa)を検出した。Okajima らの方法に基づいて SDS-PAGE を行った後、ウェスタンブロット法で解析すると、Laemmli 法によって SDS-PAGE を行ったウェスタンブロット法と比較して、HA-Cav1102-134 の検出感度が改善された(図3)。
工夫とコツ
- ゲル作製に使用する10% APS は冷蔵保存だと分解されてしまうが、冷凍保存すれば長期保存可能である。1.5 ml チューブに分注して凍結しておくと便利である。再凍結も可能だが、使い切ることを推奨する。
- 分離ゲルは固まると気泡のようなものができることがある。この場合は分離ゲル作製時に APS と TEMED を元の量の5–20%程度減らす(5)。
- PVDF メンブレンは、汚れを吸着するため、印をつけたところ以外はピンセットであってもできるだけ触らないようにする。
- 抗体の希釈濃度は組織ホモジネートサンプルや培養細胞ライセートなどで予備実験を行い、事前に条件を検討しておくとよい。
- リン酸化タンパク質を検出する場合はリン酸バッファーやスキムミルク(リン酸化されたカゼインを多く含む)の使用を避ける。
実験の安全
- アクリルアミドは神経毒性があるため、ゲル作製時には使い捨て手袋を手にはめてゲル溶液を作製する。アクリルアミドはゲル化すると毒性はなくなる。
- 電気泳動中は、感電を防ぐため電極や泳動バッファーには触れないように注意する。
文献
- Laemmli, UK., Nature, 227, 680–685 (1970)
- Okajima, T., et al., Anal. Biochem., 211, 293–300 (1993)
- Moritz, CP., J. Proteomics, 212, 103575 (2020)
- Hagiwara et al., Biochem. Biophys. Res. Commun., 378, 73–78 (2009)
- アトー株式会社ホームページ実験のコツダウンロード,PAGE 初めての電気泳動―タンパク質のポリアクリルアミドゲル電気泳動編―,https://www.atto.co.jp/content/download/6197/81407/file/(2020年9月24日閲覧)
謝辞
本研究で使用した Cav1102-134 in pCMV-HA ベクターは東京農業大学応用生物科学部農芸化学科、山本祐司教授より供与していただきました。この場を借りて深く感謝申し上げます。
-
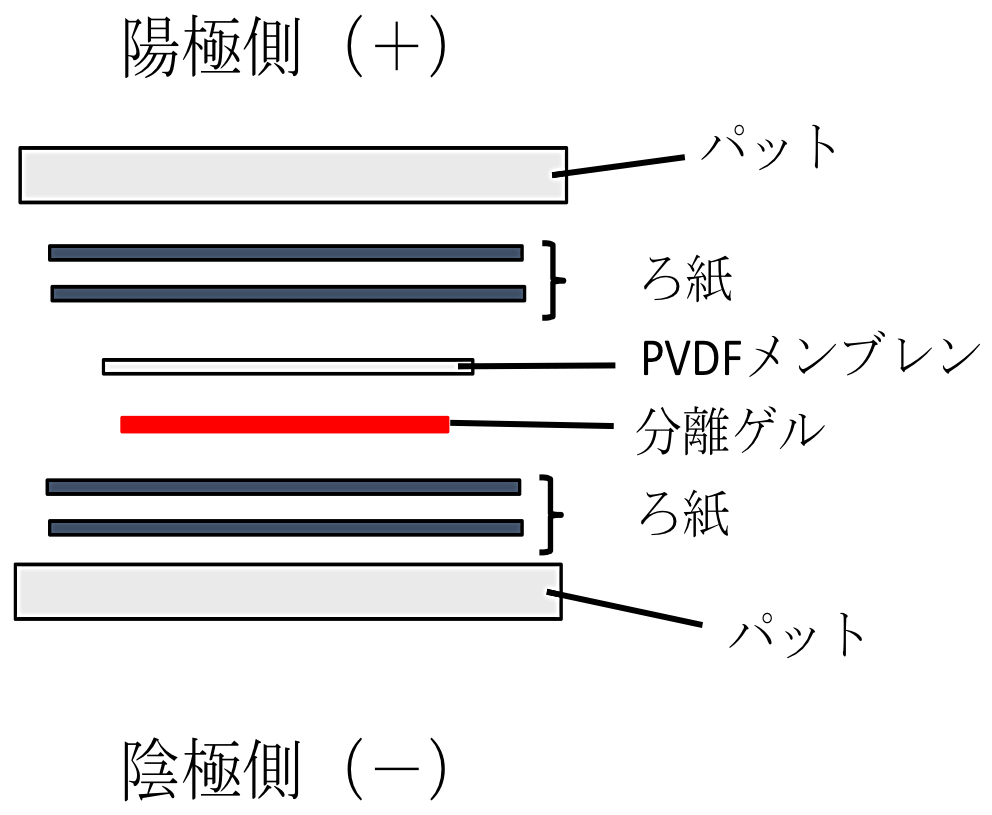
図1:ブロッティング。陰極側からパット、ろ紙2枚、分離ゲル、PVDF メンブレン、ろ紙2枚、パットの順に重ねる。 -
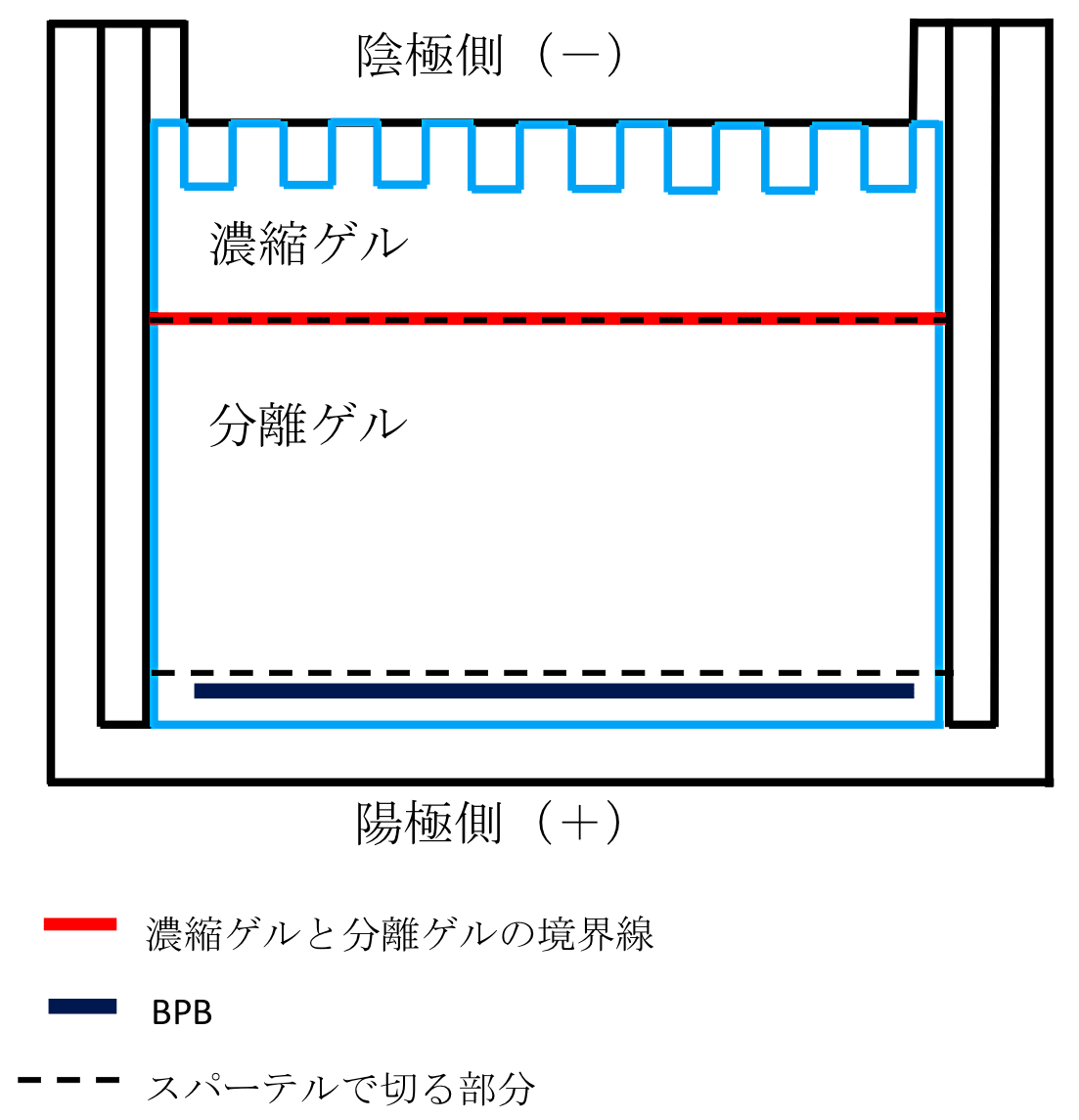
図2:ブロッティング前のゲルの切り方。ゲル板(MAB-10)とゲル板(MB-00)の間にスパーテルのへらになっている方を入れ、てこの原理でゲル板をはずす。濃縮ゲルを分離ゲルから切り離すため、スパーテルのへら側を濃縮ゲルと分離ゲルの境界線に沿って押し付け突き刺し、ゲルからスパーテルを抜き、横にずらしては突き刺し抜くといった操作を繰り返し、端から端まで切り、完全に濃縮ゲルを切り離す(スパーテルを押し付けて引いて切ろうとするとゲルが崩れるため押して切る)。また、分離ゲルの泳動先端の BPB を含めた下(陽極側)の分離ゲルも不要なため、BPB から少しだけ上(陰極側)を同様な方法でスパーテルを用いて、泳動方向に対して直角に近い角度で端から端まで切り、捨てる。ゲル板と残った分離ゲルの間にスパーテルを入れ、ゲル板を逆さまにしてあらかじめ準備しておいたゲルホルダーのろ紙の上に載せる(図1参照)。 -

図3:低分子量タンパク質検出の一例。Okajima らの方法で SDS-PAGE を行った後、ウェスタンブロット法を行ったところ、Laemmli 法と比較して、HA-Cav1102-134(5.9 kDa)のシグナルが強く検出され、低分子量タンパク質の検出感度が改善された。なお、Okajima らの方法と Laemmli 法で SDS-PAGE を行った後、転写の条件を揃えるために、それぞれのゲルのタンパク質をアプライしてないレーンを切って除き、両方法で SDS-PAGE を行ったタンパク質を同一の PVDF メンブレンに転写しており、露光時間は同じである。 -
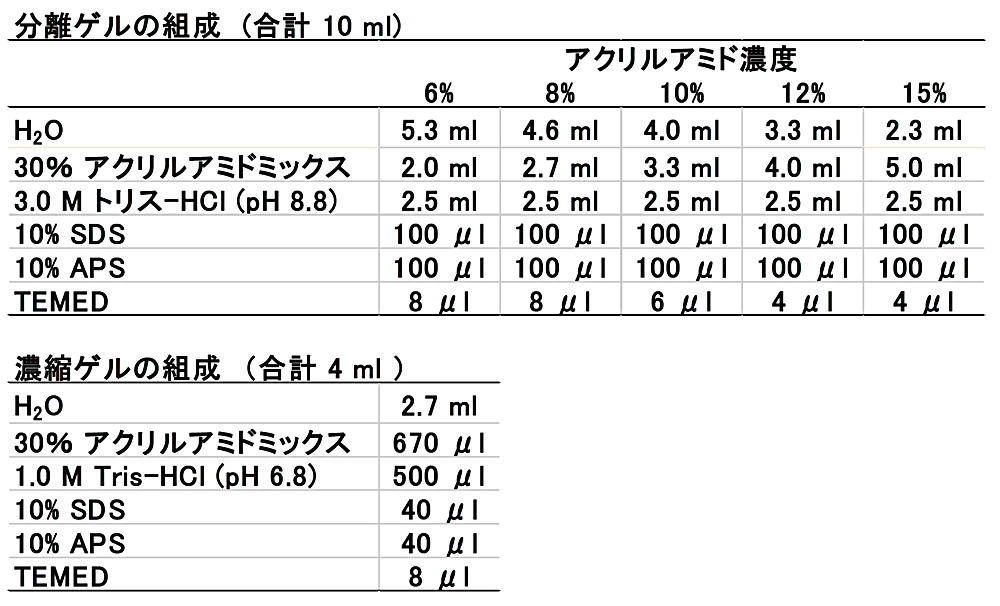
表1:ゲルの組成(Okajima らの方法(2)) -
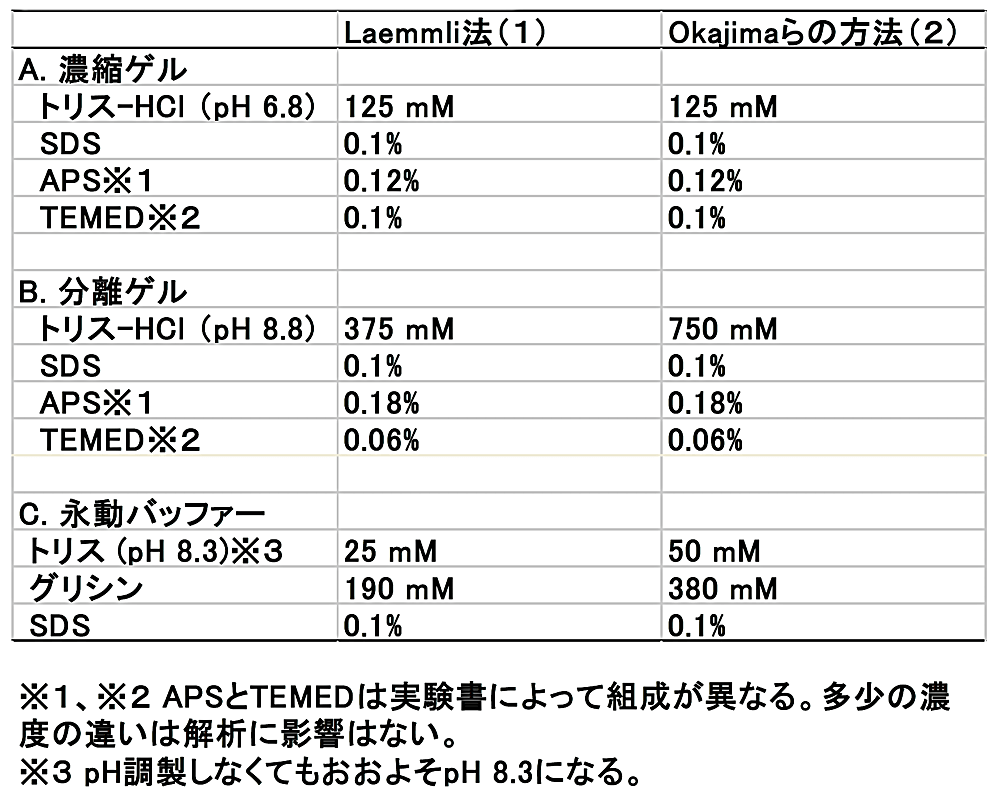
表2:各方法による試薬濃度の違い
概要
ウェスタンブロット法は、SDS-PAGE 法でタンパク質を分子量の違いによって分離した後、PVDF メンブレンなどにタンパク質を転写し、検出したいタンパク質の特異的抗体を用いて、タンパク質を可視化する方法である。研究室でよく用いられているウェスタンブロット法は、1970年に Laemmli によって報告された条件(1)で SDS-PAGE を行った後に、転写を行うことが多い。しかし、分子量が 15 kDa 以下のタンパク質は検出しづらくなるといったことが起こる。Okajima らは分離ゲルのトリス濃度や SDS-PAGE における泳動バッファーのトリス濃度とグリシン濃度について検討し、低分子量タンパク質のバンドがクリアになることを CBB 染色で報告している(2)。この方法で SDS-PAGE を行った後、ウェスタンブロット法を行うことで低分子量タンパク質のバンドがクリアになり、上述の問題点が改善される。ここでは、グリシンとトリスを使用した Okajima らの方法に基づいたウェスタンブロット法について記述する。
イントロダクション
ウェスタンブロット法(別名イムノブロット法)は、約40年前に報告された方法であるが、現在においても生化学分野や分子生物学分野をはじめとした実験系の研究室において最も頻繁に使用されているタンパク質の解析方法の一つである(3)。
ウェスタンブロット法は、SDS-PAGE でタンパク質を分子量の違いによって分離した後、PVDF メンブレンなどにタンパク質を移し、検出するタンパク質の特異的抗体を用いて、タンパク質を可視化する方法である。しかし、ウェスタンブロット法において、おおよそ 15 kDa 以下の低分子量タンパク質は、15 kDa 以上のタンパク質と比較して、検出しづらくなるといったことが起こる。補足すると低分子量タンパク質だけではなく 100 kDa 以上の高分子量タンパク質でも検出しづらくなるといったことが起こるが、高分子量タンパク質の検出方法は別の論文や著書を参考にしていただきたい。転写バッファー中のメタノール濃度を高めると転写効率が上がる傾向にあり、低分子量タンパク質を検出しやすくなる。通常は、PVDF メンブレンでは転写バッファー中のメタノール濃度を15%以下、ニトロセルロースメンブレンでは転写バッファー中のメタノール濃度を20%以下にして転写を行うことが多い。また、高分子量タンパク質では、転写効率が下がるため、ゲルからタンパク質を溶出させやすくするために転写バッファー中に SDS を0.1%以下の濃度で加えることがあるが、低分子量タンパク質では SDS を加えないほうがメンブレンへの吸着力が良い。さらに、メンブレンの種類別に比較すると、ニトロセルロースメンブレンはバックグラウンドが抑えられ、きれいなバンドが得られやすいが、タンパク質の結合能力が弱く、低分子量タンパク質は検出しにくい。従って、低分子量タンパク質を検出する際は、ニトロセルロースメンブレンよりもタンパク質の結合能力が高い PVDF メンブレンを選択したほうが良い。また、PVDF メンブレンとニトロセルロースメンブレンともに、孔径が異なったものが販売されており、孔径が 0.45 μm、0.2 μm、0.1 μm などのメンブレンがある。低分子量タンパク質は、孔径が小さいほうが、転写においてメンブレンに吸着しやすく、メンブレンをすり抜けにくくなるので、低分子量タンパク質を解析するためには、孔径の小さいメンブレンを選ぶと良い。ただし、孔径の小さいメンブレンはバックグラウンドが上がりやすいことを念頭に置く必要がある。
低分子量タンパク質を解析するための SDS-PAGE は、トリシンを使用する方法もあるが、グリシンと比較してトリシンは高価であることや、グリシンを使用した SDS-PAGE と比較してトリシンを使用した SDS-PAGE は解析に時間を要する。Okajima ら(2)は分離ゲルのトリス濃度や SDS-PAGE における泳動バッファーのトリス濃度とグリシン濃度について検討し、Laemmli によって報告された条件(1)と比較して低分子量タンパク質のバンドがクリアになることを CBB 染色で報告している。これまで、上記に記載した転写バッファー中の試薬濃度の調節や、使用するメンブレン自体を検討することによって、ウェスタンブロット法における低分子量タンパク質の検出効率を改善できることが知られていたが、筆者は、Okajima らの方法に基づいて SDS-PAGE を行い、ウェスタンブロット法で解析したところ低分子量タンパク質の検出効率が改善された。すなわち、この結果は、SDS-PAGE における分離ゲル中のトリス濃度や泳動バッファー中のトリス濃度やグリシン濃度を調節することによっても低分子量タンパク質の検出効率が改善することができることを示すものである。本論文では、低分子量タンパク質を解析するための、トリシンを使用しない、グリシンとトリスを使用した Okajima らに基づく SDS-PAGE を用いたウェスタンブロット法について記述する。
実験器具・装置
- ゲル板(泳動用ガラスプレート MAB-10 と MB-00)(アトー)
- シールガスケット(RMS-01)(アトー)
- ラピダスマグネクリップミニ(製品番号:2398239)(アトー)2個。ゲル板が固定できれば市販の大きめのダブルクリップでもよい。市販のダブルクリップを使用する場合は4個必要。
- コーム(RM 10-12)(アトー)
- 電気泳動装置(泳動槽)(AE-6500)(アトー)
- パワーサプライ(各社から販売されているもの)
- リード線
- 50 µl もしくは 25 µl のハミルトンマイクロシリンジ(筆者は25 µl のハミルトンマイクロシリンジ 702SNR<80465>を使用)
- スパーテル
- ピンセット
- ディッシュ(プラスチック製)
- 転写準備のための容器(横 18 cm、縦 26.5 cm、高さ 5.5 cm)
- 転写装置(BE-351W)(バイオクラフト)
- 振とう機
- 発光撮影装置
試薬
- H2O(純水)
- 30%アクリルアミドミックス(29%アクリルアミド、1% N,N’-メチレンビス(アクリルアミド))
- 3.0 M トリス-HCl(pH 8.8)
- 1.0 M トリス-HCl(pH 6.8)
- 10% SDS(Sodium dodecyl sulfate)
- 10% APS(Ammonium persulfate)(小分けにして凍結保存しておくと便利)
- TEMED (N,N,N’,N’-テトラメチルエチレンジアミン)
- エタノール
- 4×SDS-PAGE サンプルバッファー(400 mM トリス HCl(pH 6.8)、8% SDS、40% グリセロール、24% 2-メルカプトエタノール、0.1% ブロモフェノールブルー)
- 泳動バッファー(50 mM トリス、380 mM グリシン、0.1% SDS)(2×泳動バッファーを作製しておくと便利)
- メタノール
- パラフィルム
- 転写バッファー(20 mM トリス、150 mM グリシン)(10×転写バッファーを作製しておくと便利)
- 転写用ろ紙(CB-09A 85×90 mm)(アトー)
- PVDF メンブレン(イモビロン-P、0.45 µm、IPVH00010)(ミリポア)、あらかじめ分離ゲルと同じぐらいの大きさに切っておく。
- TBST(140 mM 塩化ナトリウム、25 mM トリス-HCl(pH 7.4)、0.05% Tween20)または、PBST(140 mM 塩化ナトリウム、8.1 mM リン酸水素二ナトリウム、2.7 mM 塩化カリウム、0.05% Tween20)
- スキムミルク、BSA などのブロッキング剤または市販のブロッキング剤
- 1次抗体
- 2次抗体(HRP 標識)
- 化学発光検出試薬
実験手順
- ゲルの作成
- サンプルの作製
- SDS-PAGE
- ウェスタンブロット法
実験の詳細
1. ゲルの作成
Okajima らの方法(2)によるゲル作製のために必要な試薬の量を、表1に示した。低分子量タンパク質を分離するためには、15%程度かそれ以上のアクリルアミドゲルで解析することになるが、参考までに6%–12%のゲルの組成についても表1に記載した。また、表2には Laemmli 法(1)、Okajima らの方法(2)における試薬の濃度を記載した。Laemmli 法の分離ゲルと比べて、Okajima らの方法の分離ゲルはトリス-HCl(pH 8.8)の濃度が2倍である。以下に手順を記載する。
ゲル板の汚れを取り除くために、ゲル板の内側に70%エタノールを吹き付けて、キムワイプで拭く。ゲル板(MAB-10)にシールガスケットをはめこみ、ゲル板(MB-00)を重ねて、ラピダスマグネクリップミニでゲル板を固定する。
表1の分離ゲルの組成に従い、H2O、30%アクリルアミドミックス、3.0 M トリス-HCl(pH 8.8)、10% SDS、10% APS、TEMED の順に15 ml チューブに加え、転倒混和する。調製した分離ゲル溶液 5.5 ml(アトーの泳動用ガラスプレート MAB-10 と MB-00 の場合)をゲル板に流し込む。そして、分離ゲルの上にエタノールを重層し、分離ゲルが固まるまで静置する(30分から60分間)。分離ゲルが固まったら、濃縮ゲルを作製するために、表1の濃縮ゲルの組成に従い、H2O、30%アクリルアミドミックス、1.0 M トリス-HCl(pH 6.8)、10% SDS、10% APS、TEMED の順に 15 ml チューブに加え、転倒混和する。ゲル板を逆さまにして、分離ゲルの上に重層したエタノールを捨てる。調製した濃縮ゲル溶液をゲル板の一番上まで流し込み、コームを差し込み、濃縮ゲルが固まるまで静置する(30分から60分間)。ゲルは1日程度なら、ラップで包んで冷蔵保存可能である。
2. サンプルの調製
組織や培養細胞からタンパク質を抽出したサンプルあるいは精製したタンパク質などサンプルのタンパク濃度を測定し、解析するタンパク質量を見積もる。サンプルに適宜 H2O を加え、液量を調節し、サンプルと4×サンプルバッファーを 3 : 1 の割合で混合し(1.5 ml チューブを使用)、キャップロックでチューブのふたを留め、ヒートブロックを用いて95 ℃で5分間加熱する。この加熱によりタンパク質の高次構造が壊れる。加熱後すぐにキャップロックを取り外すと、加熱によって蒸発した水によって、チューブ内部の圧力の上昇により、1.5ml チューブのふたが開いてしまうことがあるため、サンプルが少し冷えるまで待つ。加熱後は短時間の遠心(フラッシュ)によってサンプル溶液全量をチューブの下側に集める。
3. SDS-PAGE
電気泳動装置の下側に泳動バッファーを加える。1. で作成したゲルのゲル板からクリップ、シールガスケット、コームをはずし、ゲル板を電気泳動装置にセットする。ゲル板の下側に空気が入っていたら電気泳動装置を斜めにして空気を抜く。電気泳動装置の上側に泳動バッファーを加える。ハミルトンシリンジの針がゲルに突き刺さらないようウェル内に入れ、ハミルトンシリンジで泳動バッファーを吸って勢いよく出す操作を何度も繰り返し、ウェル内の未重合のアクリルアミドやゲルの破片を取り除く。ハミルトンシリンジを用いて各サンプルを別々のウェルに入れていく。また、分子量マーカーを入れるウェルを残しておき、そのウェルに分子量マーカーを加える。電気泳動装置とパワーサプライをリード線でつなぎ、20 mA(定電流)で泳動する。BPB がゲルの先端近くまで移動したら、電源を切る。なお、SDS-PAGE を行っている間に、PVDF メンブレンの親水処理を行っておく(下記のウェスタンブロット法の手順に記載)。
4. ウェスタンブロット法
SDS-PAGE を行っている間に、10×転写バッファーを希釈し、1×転写バッファー 1 ℓ の調製と PVDF メンブレンの浸水処理を行う。ピンセットで、PVDF メンブレンの右端上の角を掴んで、ディッシュに移す(左利きの人は左端上の角でよい)。油性ダーマトグラフで右端上の角に印をつける(表裏を区別するためである)。以後できるだけ印をつけた部分をピンセットで掴むようにする。PVDF メンブレンに、メタノールが浸るまで加えて、よく馴染ませて、1分間静置する。メタノールを除き、転写バッファーを PVDF メンブレンが浸るまで加える。PVDF メンブレンが転写バッファーをはじき、浮き上がってくることがあるので、PVDF メンブレンと同じぐらいの大きさに切ったパラフィルムをかぶせて、PVDF メンブレンを転写バッファーに馴染ませ、SDS-PAGE が終わるまで静置しておく。PVDF メンブレンは以後乾かさないようにする。
SDS-PAGE が終了したら、転写の準備に移る。容器(横 18 cm、縦 26.5 cm、高さ 5.5 cm)にディッシュに加えなかった残りの転写バッファーを全量移し、パット(スポンジ)とろ紙を転写バッファーに入れ、手袋をはめた手で押し空気を抜く。マイナス(-)極側からパット、ろ紙(2枚)、分離ゲル、PVDF メンブレン、ろ紙(2枚)、パットの順にゲルホルダーに載せていき(図1)、すべて載せたらゲルホルダーで挟む。このときに、分離ゲルと PVDF メンブレンの間に空気が入っているとその部分の転写がうまくいかないので、空気が入っている場合は、プラス(+)極側のろ紙とパットをかぶせる前に、PVDF メンブレンと分離ゲルを手袋をはめた手で押して空気を除く。なお、SDS-PAGE 終了後のゲルの切り方については図2に示したので参考にしていただきたい。ゲルホルダーの+と-の向きを間違えないように転写装置に移し、容器とディッシュの転写バッファー全量を転写装置に加えた後、転写装置とパワーサプライをリード線でつなぎ、100 V の定電圧で1~2時間通電する。
転写している間に、ブロッキングのための5%スキムミルク溶液または5% BSA 溶液を TBST か PBST で 25 ml 調製しておく。なお、リン酸化タンパク質を検出する場合は、BSA を TBST で希釈したものをブロッキング剤として使用する(工夫とコツを参照)。また、市販のブロッキング剤を使用することで、検出時のバックグラウンドが軽減されることもある。転写が終了したら、ゲルホルダーを転写装置から取り出した後、ピンセットで PVDF メンブレンをディッシュに移し、ブロッキング溶液を加えて、振とう機を用いて60分間以上室温で振とうし、ブロッキングする(冷蔵庫で一晩反応させることも可能である)。ブロッキング終了後、ブロッキング剤を捨て、TBST もしくは PBST を PVDF の入っているディッシュに加えて、振とう機で5分間振とうして洗浄する。洗浄液を捨て、同様の操作でさらに2回以上洗浄する。
ピンセットで PVDF メンブレンを乾いたディッシュに移し、TBST または PBST で希釈した1次抗体(上記に記載したように、リン酸化タンパク質を検出する抗体の場合は TBST で希釈する)をピペットで PVDF メンブレンの上に垂らし、ディッシュを少し傾けるなどしてよく馴染ませる。抗体の使用量を節約する場合は少量の抗体希釈液を PVDF メンブレンに垂らし、よく馴染ませて、ディッシュを傾けて抗体溶液をディッシュの端に集めて、ピペットで吸って、その溶液を再び PVDF メンブレンに垂らし、よく馴染ませるといった操作を繰り返す。その後、PVDF メンブレンの上に PVDF メンブレンより少し大きめのパラフィルムを載せ、空気を抜きぴったりと密着させる。室温で1時間から2時間程度静置する(冷蔵庫で一晩反応も可能)。抗体反応終了後、TBST もしくは PBST をディッシュに加えて、振とう機で5分間振とうして PVDF メンブレンを洗浄する。洗浄溶液を捨て、同様の操作でさらに2回以上洗浄する。
ピンセットで PVDF メンブレンを乾いたディッシュに移し、TBST または PBST で希釈した2次抗体を PVDF メンブレンの上に垂らし、ディッシュを少し傾かせるなどしてよく馴染ませる。抗体の使用量を節約する場合は一時抗体の時と同様の操作を行うと良い。その後、PVDF メンブレンの上に PVDF メンブレンより少し大きめのパラフィルムを載せ、空気を抜きぴったりと密着させる。室温で1時間静置する。2次抗体の反応時間が長くなると検出時にバックグラウンド上がるためタイマーで時間を計っておいたほうが良い。抗体反応終了後、TBST もしくは PBST をディッシュに加えて、振とう機で5分間振とうして PVDF メンブレンを洗浄する。洗浄溶液を捨て、同様の操作でさらに2回以上洗浄する。
洗浄終了後、PVDF メンブレンの洗浄溶液をよく切る。PVDF メンブレンの上にピペットを用いて発光試薬(筆者は Wako のイムノスター LD を使用)を垂らし、ピンセットで PVDF メンブレンを持ち上げ傾かせるなどしてよく馴染ませる。その後、発光撮影装置で、タンパク質を検出する。
実験例
脂質ラフトの一種であるカベオラに局在するタンパク質である caveolin-1 の N 末端側より102番目から134番目のアミノ酸配列(Cav1102-134)を導入した pCMV-HA ベクター(Cav1102-134 in pCMV-HA ベクター)(4)を子宮頸癌細胞である HeLa 細胞にトランスフェクションし、48時間培養した。細胞を回収し、UltraRIPA kit for Lipid Raft(BioDynamics Laboratory Inc.)を用いて、脂質ラフトタンパク質を抽出し、上記の方法でウェスタンブロット法を行った。
転写の条件を揃えるため、SDS-PAGE 後にタンパク質をアプライしていない余分なレーンはゲルを切って除き、Okajima らの方法で SDS-PAGE を行ったゲルに含まれるタンパク質と Laemmli 法によって SDS-PAGE を行ったゲルに含まれるタンパク質を同じ PVDF メンブレンに転写した。ブロッキング剤は、PVDF Blocking Reagent for Can Get Signal(TOYOBO)を使用しブロッキングを行った。1次抗体は抗 HA マウスモノクローナル抗体(10,000倍希釈)(SIGMA-ALDRICH H9658)、2次抗体は HRP 標識された抗マウス IgG (IBL #17601)を使用し、化学発光法で HA-Cav1102-134(約 5.9 kDa)を検出した。Okajima らの方法に基づいて SDS-PAGE を行った後、ウェスタンブロット法で解析すると、Laemmli 法によって SDS-PAGE を行ったウェスタンブロット法と比較して、HA-Cav1102-134 の検出感度が改善された(図3)。
工夫とコツ
- ゲル作製に使用する10% APS は冷蔵保存だと分解されてしまうが、冷凍保存すれば長期保存可能である。1.5 ml チューブに分注して凍結しておくと便利である。再凍結も可能だが、使い切ることを推奨する。
- 分離ゲルは固まると気泡のようなものができることがある。この場合は分離ゲル作製時に APS と TEMED を元の量の5–20%程度減らす(5)。
- PVDF メンブレンは、汚れを吸着するため、印をつけたところ以外はピンセットであってもできるだけ触らないようにする。
- 抗体の希釈濃度は組織ホモジネートサンプルや培養細胞ライセートなどで予備実験を行い、事前に条件を検討しておくとよい。
- リン酸化タンパク質を検出する場合はリン酸バッファーやスキムミルク(リン酸化されたカゼインを多く含む)の使用を避ける。
実験の安全
- アクリルアミドは神経毒性があるため、ゲル作製時には使い捨て手袋を手にはめてゲル溶液を作製する。アクリルアミドはゲル化すると毒性はなくなる。
- 電気泳動中は、感電を防ぐため電極や泳動バッファーには触れないように注意する。
文献
- Laemmli, UK., Nature, 227, 680–685 (1970)
- Okajima, T., et al., Anal. Biochem., 211, 293–300 (1993)
- Moritz, CP., J. Proteomics, 212, 103575 (2020)
- Hagiwara et al., Biochem. Biophys. Res. Commun., 378, 73–78 (2009)
- アトー株式会社ホームページ実験のコツダウンロード,PAGE 初めての電気泳動―タンパク質のポリアクリルアミドゲル電気泳動編―,https://www.atto.co.jp/content/download/6197/81407/file/(2020年9月24日閲覧)
謝辞
本研究で使用した Cav1102-134 in pCMV-HA ベクターは東京農業大学応用生物科学部農芸化学科、山本祐司教授より供与していただきました。この場を借りて深く感謝申し上げます。
