

概要
抗体は研究試薬として幅広く用いられるが、抗体の性能がロットにより異なり、また抗体の性能を評価する統一基準が未だ確立されていない。そのため市販抗体の多くにおいて性能評価が不十分で、研究者自身が使用する抗体を評価しなければならないのが現実である。抗体の一般的な評価法として、ドットブロットやペプチドアレイ法が用いられているが、実験条件により結果が大きく異なり、抗体の特異性を絶対的な基準のもと評価できない問題点があった。抗体の特異性は、標的抗原(On target)及び非標的抗原(Off targets)それぞれへの親和性の違いにより定義されるため、抗体親和性の指標である平衡解離定数(KD 値)を決定することができれば、On target 及び Off targets への KD 値の違いから抗体の特異性を定量的に評価できる。一方、抗体の平衡解離定数を決定する物理化学的な解析手法においては、多量の抗体を必要とするため、高価な市販抗体の定量的特異性評価には適していない。そこで著者らは、1 μg というわずかな抗体量で複数の抗原に対する見かけのKD 値を決定し、抗体の特異性を定量的に評価できる Peptide IP assay を開発した。本稿では、Peptide IP assay の特徴と詳細な手順について、抗ヒストン翻訳後修飾抗体の特異性評価を例に挙げ紹介する。
イントロダクション
近年、研究試薬として用いられる抗体の特異性の低さやロット間での特異性の違いが問題となっている(1)。研究試薬抗体のほとんどは、血清やポリクローナル抗体であるため、カタログ番号上は同一であっても、ロットにより抗体の性能が異なる。また、実際には性能が低いにもかかわらず、研究者が購入した抗体を特異性が高いものとして使用していることも大きな問題となっている。特にエピジェネティクス分野においては、抗体を用いた実験手法が重要な役割を果たしており、市販抗体の特異性問題が、信頼性・再現性の高い結果を取得する上で障害となっている(2-5)。購入した抗体のロットがデータシートに記載されているロットと異なる場合も多く、研究者は購入した抗体の特異性を自身で評価する必要がある。一方で、高価な市販抗体を浪費せずにその特異性を評価する手法には限りがあり、研究者が抗体の性能を統一基準のもと評価できない課題があった。
ウェスタンブロットやドットブロット、ペプチドアレイ法は、簡便であるため一般的な抗体特異性評価法として広く用いられているが、これらはバンドやスポットの濃淡を比較する定性的な評価法であり、さらに抗体濃度や洗浄条件などによって見かけの特異性が大きく異なるという根本的な問題がある。抗体の特異性を定量的に評価する方法として、Ontarget 及び Off targets に対する抗体の KD 値を決定し、比較する手法がある。しかし、等温滴定型熱量測定法(ITC)や表面プラズモン共鳴法(SPR)など KD 値を決定できる一般的な手法は、多くの抗体量を必要とし、購入した抗体を目的実験に使用する前に使い切ってしまう問題点があった。
これらの問題を踏まえ、著者らは、わずかな抗体量で抗体の特異性を定量的に評価できる Peptide IP assay を開発した(図1)(3)。Peptide IP assay では、ビーズに固定化した抗体と抗原との相互作用を蛍光検出により測定する。抗体が捕捉した抗原の量に比例して蛍光強度が強くなるため、抗原濃度に対する蛍光強度をプロットすることで “見かけ” の KD 値を決定できる。ビーズを酵母と見立てると、Peptide IP assay は酵母表層提示法による抗体の見かけの平衡解離定数決定法に類似しており、この手法で決定された見かけの KD 値は、物理化学的な手法で算出された抗体の KD 値とほとんど差がないことも報告されている(6)。
Peptide IP assay は、主に免疫沈降実験に使う抗体の特異性評価のために開発した手法である。エピジェネティクス分野においてクロマチン免疫沈降と次世代シーケンサーを組み合わせた ChIP-seq 法は、ゲノム解析に必須の手法であるが、一回の実験で得られるデータ量と結果のインパクトが大きいため、より正確な実験精度が求められる。そのため、ChIP 実験前に使用する抗体の特異性を厳しく評価することが重要であるが、ドットブロットやペプチドアレイおいて高い特異性を示した抗体が、ChIP 実験において機能しない例が報告されている(2)。この現象は、目的実験と抗体評価法のフォーマットのミスマッチが原因であると考えられる。すなわち、“ブロット” タイプの評価法は、支持担体に高密度に固定化された抗原と相互作用する抗体を検出するフォーマットである一方、“免疫沈降” タイプの実験では、支持担体に抗体を固定化し、抗体と相互作用した抗原を検出もしくは測定する。Peptide IP assay は、免疫沈降を模倣したフォーマットであるため、ChIP に使用する抗体の特異性評価に最適である。実際、Peptide IP assay において高い特異性を示した抗体が ChIP-seq においても高い性能で機能している(4)。
本稿では、Peptide IP assay を用いて抗ヒストン翻訳後修飾抗体の特異性を評価する手順を紹介する。本手法はヒストンペプチドのみならず、ビオチン化したタンパク質やペプチドを抗原として用いることも可能であり、様々な抗体の特異性の定量的評価にも応用できるため、汎用性の高い抗体の性能評価法となり得る。
装置・器具・試薬
- フローサイトメーター(各社)
- マイクロチューブ用ローテーター(各社)
- プレートシェイカー(各社)
- ボルテックスミキサー(各社)
- 磁気ビーズスタンド(各社)
- 96well プレート(各社)
- 96well フィルタープレート(MultiScreenHTS HV, Millipore)
- マルチスクリーンバキュームマニフォールド(Millipore)
- Protein A or Protein G Dynabeads(Thermo)
- ビオチン化ペプチド(各社。著者らは、HPLC により精製した純度95%以上のペプチドを使用している。ヒストンペプチド配列は表1を参照すること。Cys 残基を利用したビオチンの化学修飾法については、文献3を参照すること)
- 蛍光標識ストレプトアビジン(各社)
実験手順
I マグネティックビーズへの抗体の固定化
- ビーズの準備
- ビーズと抗体の混合及び洗浄
II 抗体–抗原反応
- ビオチン化ペプチドの準備
- 抗体固定化ビーズとペプチドの混合及び洗浄
- 検出試薬の添加と洗浄
III フローサイトメトリーによる蛍光の検出
- 測定の準備
- サンプルの測定
IV データ解析と平衡解離定数(KD)値の算出
実験の詳細
I マグネティックビーズへの抗体の固定化
Peptide IP assay では、抗体–抗原反応中に抗体がビーズから解離することを防ぐため、抗体はビーズに強固に固定化されている必要がある。IgG 結合タンパク質である Protein A 及び Protein G の抗体への親和性は、動物種とサブクラスによって異なるため、実験者は使用する抗体に高い親和性を示す IgG 結合タンパク質を選択しなくてはならない。例えばマウス由来の抗体には Protein G ビーズが適しており、ラビット由来の抗体には Protein A ビーズが適している。本稿では、Protein A ビーズを用いた実験例を記す(3,4)。
1) ビーズの準備
Protein A Dynabeads をボルテックスミキサーでよく懸濁し、10 μL のビーズを100 μL のPBST/BSA [PBS, 0.5% (W/V) BSA, 0.1% (V/V) Tween20] 溶液に加え、ピペッティングにより攪拌する。マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。この操作をもう一度繰り返す。
2) ビーズと抗体の混合及び洗浄
1 μg の抗体を100 μL の PBST/BSA 溶液に添加し、ピペッティングまたはタッピングにより攪拌混合することで抗体希釈液を作る。抗体希釈溶液をステップ1で調製したビーズに添加し、4℃にて1時間、ローテーターを用いて攪拌混合する。1ビーズあたりの抗体固定化量が不均一となるため、抗体原液をビーズ懸濁液に直接添加することは避ける。攪拌混合後、マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。300 μL の PBST/BSA 溶液でビーズを懸濁し、マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。この操作をもう一度繰り返し、最後に抗体固定化ビーズを100 μL の PBST/BSA 溶液に懸濁する。抗体固定化ビーズは、ヒストンペプチドと混合する直前に PBST/BSA 溶液にて40倍希釈する。
II 抗体–抗原反応
抗体を固定化したビーズとビオチン化ペプチドを混合し、抗体–抗原結合反応を行う。抗原結合反応が平衡に達した後、抗体に未結合のペプチドを洗浄し、蛍光標識ストレプトアビジンを加える。ビーズ上の抗体と相互作用しているビオチン化ペプチドに蛍光標識ストレプトアビジンが結合することで、フローサイトメトリーにより抗体–抗原相互作用に依存した蛍光を検出することが可能となる(図1)。
1) ビオチン化ペプチドの準備
各抗体–抗原反応において、抗体固定化ビーズと等量のビオチン化ペプチドを混合するため、実験者は測定したい濃度の2倍濃いビオチン化ペプチドを準備する。平衡解離定数 KD を算出するため結合曲線を描きたい場合、複数のデータポイントが必要となるため、実験者はあらかじめ連続希釈した溶液を作製しておく。ペプチド濃度の決定については、「工夫とコツ」を参照すること。希釈液は PBST/BSA である。
2) 抗体固定化ビーズとペプチドの混合及び洗浄
10 μL の希釈した抗体固定化ビーズと10 μL のビオチン化ペプチドを混合する。サンプル数が多い場合は、96well プレートを用いると便利である。抗体の親和性が著しく高く(例えば抗体の KD 値が picomolar 領域) 、抗原ペプチド濃度を低くする必要がある場合には、抗体固定化ビーズの量を減らす、もしくは反応体積を増やすことで ligand depletion(抗体量に対する抗原量の不足)を防ぐことができる。4℃で30分間振盪させた後、ビーズとペプチドの混合液を96well フィルタープレートに移す。バキュームマニフォールドを用いて溶液を吸引し、300 μL の PBST/BSA を各 well に添加し吸引することで、抗体固定化ビーズを洗浄する。洗浄操作を2回繰り返す。
3) 検出試薬の添加と洗浄
20 μL の10 μg/mL 蛍光標識ストレプトアビジン(著者らは、Streptavidin Dylight650(Thermo)を使用している)を各 well に添加する。4℃で30分間振盪させた後、バキュームマニフォールドを用いて溶液を吸引し、300 μL の PBST/BSA を各 well に添加し吸引することで、抗体固定化ビーズを洗浄する。洗浄操作を2回繰り返す。
III フローサイトメトリーによる蛍光の検出
1) 測定の準備
実験者はまず、ペプチドと反応させていない抗体固定化ビーズをフローサイトメーターに流し、scatter plot にてビーズ集団にゲートをかける。Dynabeads は粒径が均一であるため、scatter plot にてビーズ集団が集約しており、ビーズを判別しやすい。その他、検出感度を調整するためのゲイン値などの設定は、装置によって異なるため、実験者が最適な値を設定する。
2) サンプルの測定
100 μL の PBST/BSA 溶液を用いてビーズを懸濁し、フローサイトメーターに流すことで抗体に結合したペプチドに由来する蛍光を検出する。ゲート内のビーズカウントが、500以上あれば信頼度の高い平均蛍光強度値(MFI: Mean Fluorescence Intensity)を得ることができる。
IV データ解析と平衡解離定数(KD)値の算出
フローサイトメーターに付属のソフトウエア、もしくは FlowJo などの解析ソフトを用いて、各サンプルの MFI を算出する。抗体の標的ペプチドに対する結合曲線は、ペプチド濃度に対して MFI をプロットすることで描くことができる。平衡解離定数(KD 値)は、下記の式-1を用い、非線形最小二乗法にてカーブフィッティングを行うことで算出することができる。
\[\mathrm{MFI}(x) = \mathrm{MFI}(0) + \{\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\} \times (\frac{x}{K_D + x}) \tag{式-1}\]ここで \(x\) はペプチドの濃度、\(\mathrm{MFI}(x)\) はあるペプチド濃度での MFI、\(\mathrm{MFI}(0)\) はペプチドが非存在下(ペプチド濃度が0)での MFI 、\(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) は飽和濃度のペプチドを加えたときの \(\mathrm{MFI}\) から \(\mathrm{MFI}(0)\) を差し引いた値である。カーブフィッティングに際しては、KD 値のみを未知数として扱い、\(\mathrm{MFI}(0)\) 及び \(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) は固定値として扱う。非標的ペプチド(Off targets)への平衡解離定数は、\(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) がすべての抗原ペプチドで等しくなると仮定してカーブフィッティングを行い、算出する。Peptide IP assay による抗体特異性評価の例として、抗ヒストン H3 トリメチル化 Lys9 抗体について結合評価を行った結果を図2に示す。
工夫とコツ
ビーズの取り扱いについて
ビーズは沈殿しやすく、いずれのステップにおいてもビーズをよく懸濁することが重要である。
ビーズの種類について
本稿では、マグネティックビーズを用いた Peptide IP assay を紹介したが、Protein A や Protein G がコーティングされているポリスチレンビーズを使用することも可能である(3,4)。ポリスチレンビーズを用いる場合、遠心によりビーズを分離する。
非特異吸着の確認
実験者は、抗体を固定化していないビーズとペプチドを混合し、ビーズが抗原ペプチドに非特異吸着しないかを確認する必要がある。著者らは、H4K20me1 ペプチドが Protein A をコーティングしてあるポリスチレンビーズ(Spherotech)に非特異的に吸着することを観測している(3)。
濃度が不明な市販抗体について
ポリクローナル抗体や血清において、抗体濃度が不明な場合がある。その場合、まず1 μL の抗体をビーズに固定化し、測定を行う。MFI が低く、結合曲線が描けない場合、a) ビーズへの抗体固定化量が少ない、b) 抗体自身の結合特性である、可能性がある。原因が前者である場合は、抗体量を増やすことで問題を解決できる。
抗原濃度域の決定について
抗体の標的抗原に対する見かけの平衡解離定数を算出したい場合、KD 値の濃度を含む抗原濃度域にて結合曲線を描く必要がある。そのため著者らは、まず標的抗原のみを用いて予備実験を行い、抗体の標的抗原に対する KD 値を見積もることを勧める。予備実験においては、数 μM の抗原を最大濃度として用い、濃度を細かく振る必要は無い。予備実験より得られた結合曲線からおおよその抗体の KD 値を見積もることができる。
本手法で算出される見かけの平衡解離定数(KD 値)について
Peptide IP assay においては、a) 抗体と Protein A/G との相互作用、b) 抗体と抗原の相互作用、c) ビオチン化ペプチドと蛍光標識ストレプトアビジンの相互作用、の3つの相互作用が存在する。IgG 結合タンパク質の抗体への結合親和性は、KD 値が~10-9 M と高く(7)、加えて avidity 効果(多価効果)により見かけの親和性はさらに高くなる。またストレプトアビジンのビオチンへの結合親和性は KD 値が~10-15 M と非常に高い。すなわち、(a) と (c) の相互作用は、(b) 抗体–抗原相互作用と比較して非常に強いため無視でき、本手法で算出される見かけの KD 値は (b) 抗体–抗原相互作用を反映していると著者らは考えている。
ビオチン化組換え抗体断片の利用
本稿では、IgG 抗体を用いた Peptide IP assay を紹介したが、ビオチン化した組換え抗体断片(Fab など)を用いることも可能である。その場合、ストレプトアビジンがビーズに固定化されている M280 Dynabeads(Thermo)を使用する。詳しくは、文献4を参照すること。
文献
- Bradbury, A. & Plückthun, A., Nature, 518, 27–9 (2015)
- Egelhofer, T.A. et al., Nat. Struct. Mol. Biol., 18, 91–3 (2011)
- Nishikori, S. et al., J. Mol. Biol., 424, 391–9 (2012)
- Hattori, T. et al., Nat. Methods, 10, 992–5 (2013)
- Rothbart, S.B. et al., Mol. Cell, 59, 502–11 (2015)
- Chao, G. et al., Nat. Protoc., 1, 755–68 (2006)
- Sidorin, E.V. & Solov’eva, T.F., Biochemistry (Moscow), 76, 295–308 (2011)
-
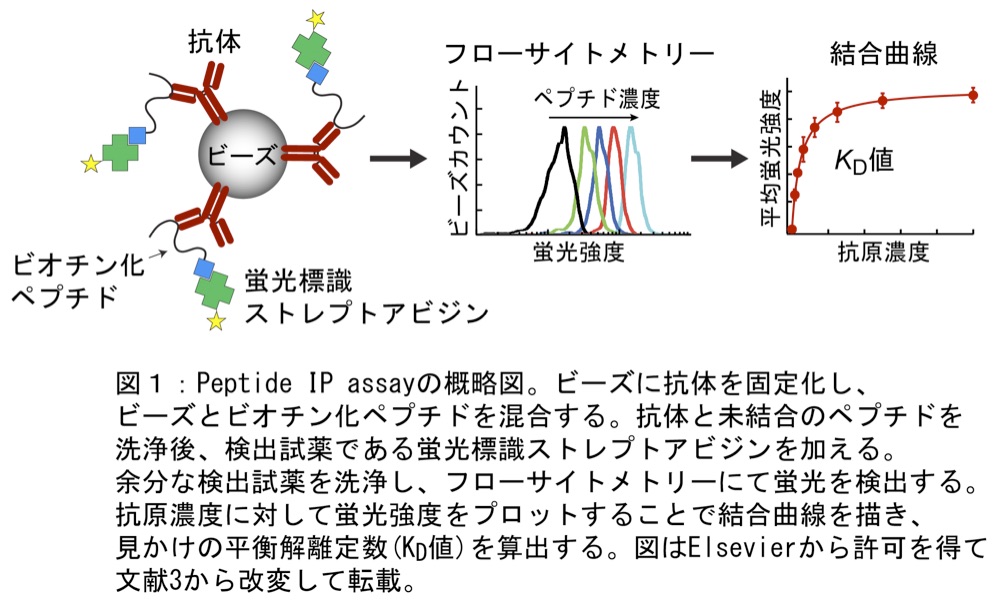
図1:Peptide IP assay の概略図 -
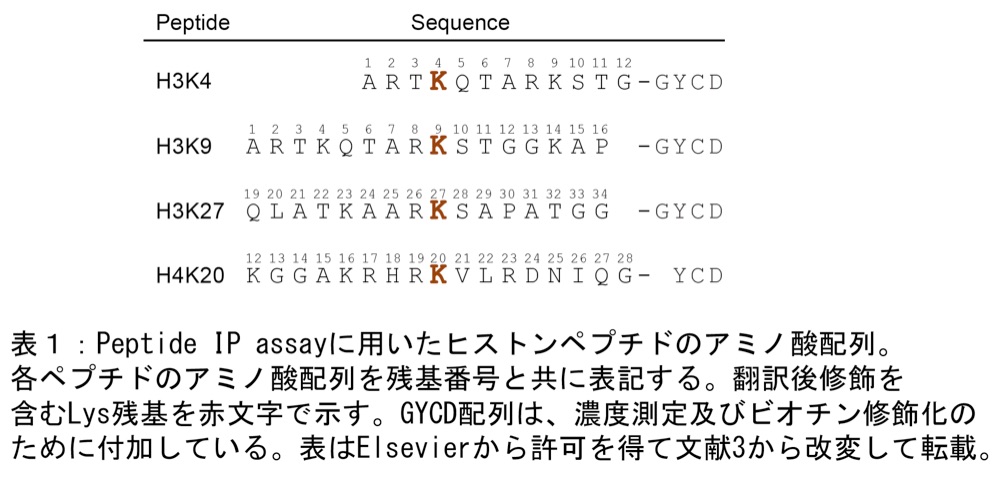
表1:Peptide IP assay に用いたヒストンペプチドのアミノ酸配列 -
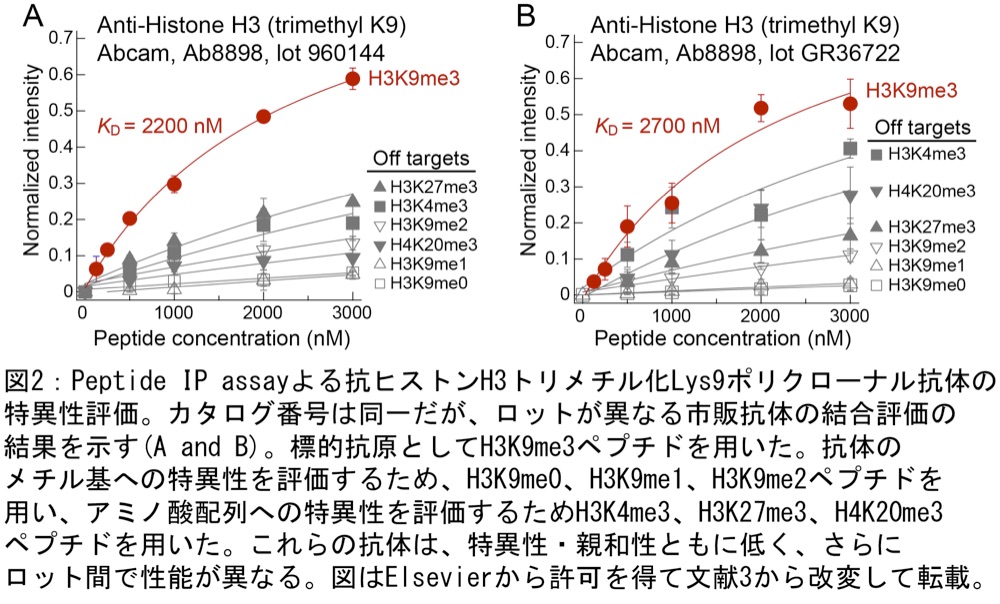
図2:Peptide IP assay よる抗ヒストン H3 トリメチル化 Lys9 ポリクローナル抗体の特異性評価
概要
抗体は研究試薬として幅広く用いられるが、抗体の性能がロットにより異なり、また抗体の性能を評価する統一基準が未だ確立されていない。そのため市販抗体の多くにおいて性能評価が不十分で、研究者自身が使用する抗体を評価しなければならないのが現実である。抗体の一般的な評価法として、ドットブロットやペプチドアレイ法が用いられているが、実験条件により結果が大きく異なり、抗体の特異性を絶対的な基準のもと評価できない問題点があった。抗体の特異性は、標的抗原(On target)及び非標的抗原(Off targets)それぞれへの親和性の違いにより定義されるため、抗体親和性の指標である平衡解離定数(KD 値)を決定することができれば、On target 及び Off targets への KD 値の違いから抗体の特異性を定量的に評価できる。一方、抗体の平衡解離定数を決定する物理化学的な解析手法においては、多量の抗体を必要とするため、高価な市販抗体の定量的特異性評価には適していない。そこで著者らは、1 μg というわずかな抗体量で複数の抗原に対する見かけのKD 値を決定し、抗体の特異性を定量的に評価できる Peptide IP assay を開発した。本稿では、Peptide IP assay の特徴と詳細な手順について、抗ヒストン翻訳後修飾抗体の特異性評価を例に挙げ紹介する。
イントロダクション
近年、研究試薬として用いられる抗体の特異性の低さやロット間での特異性の違いが問題となっている(1)。研究試薬抗体のほとんどは、血清やポリクローナル抗体であるため、カタログ番号上は同一であっても、ロットにより抗体の性能が異なる。また、実際には性能が低いにもかかわらず、研究者が購入した抗体を特異性が高いものとして使用していることも大きな問題となっている。特にエピジェネティクス分野においては、抗体を用いた実験手法が重要な役割を果たしており、市販抗体の特異性問題が、信頼性・再現性の高い結果を取得する上で障害となっている(2-5)。購入した抗体のロットがデータシートに記載されているロットと異なる場合も多く、研究者は購入した抗体の特異性を自身で評価する必要がある。一方で、高価な市販抗体を浪費せずにその特異性を評価する手法には限りがあり、研究者が抗体の性能を統一基準のもと評価できない課題があった。
ウェスタンブロットやドットブロット、ペプチドアレイ法は、簡便であるため一般的な抗体特異性評価法として広く用いられているが、これらはバンドやスポットの濃淡を比較する定性的な評価法であり、さらに抗体濃度や洗浄条件などによって見かけの特異性が大きく異なるという根本的な問題がある。抗体の特異性を定量的に評価する方法として、Ontarget 及び Off targets に対する抗体の KD 値を決定し、比較する手法がある。しかし、等温滴定型熱量測定法(ITC)や表面プラズモン共鳴法(SPR)など KD 値を決定できる一般的な手法は、多くの抗体量を必要とし、購入した抗体を目的実験に使用する前に使い切ってしまう問題点があった。
これらの問題を踏まえ、著者らは、わずかな抗体量で抗体の特異性を定量的に評価できる Peptide IP assay を開発した(図1)(3)。Peptide IP assay では、ビーズに固定化した抗体と抗原との相互作用を蛍光検出により測定する。抗体が捕捉した抗原の量に比例して蛍光強度が強くなるため、抗原濃度に対する蛍光強度をプロットすることで “見かけ” の KD 値を決定できる。ビーズを酵母と見立てると、Peptide IP assay は酵母表層提示法による抗体の見かけの平衡解離定数決定法に類似しており、この手法で決定された見かけの KD 値は、物理化学的な手法で算出された抗体の KD 値とほとんど差がないことも報告されている(6)。
Peptide IP assay は、主に免疫沈降実験に使う抗体の特異性評価のために開発した手法である。エピジェネティクス分野においてクロマチン免疫沈降と次世代シーケンサーを組み合わせた ChIP-seq 法は、ゲノム解析に必須の手法であるが、一回の実験で得られるデータ量と結果のインパクトが大きいため、より正確な実験精度が求められる。そのため、ChIP 実験前に使用する抗体の特異性を厳しく評価することが重要であるが、ドットブロットやペプチドアレイおいて高い特異性を示した抗体が、ChIP 実験において機能しない例が報告されている(2)。この現象は、目的実験と抗体評価法のフォーマットのミスマッチが原因であると考えられる。すなわち、“ブロット” タイプの評価法は、支持担体に高密度に固定化された抗原と相互作用する抗体を検出するフォーマットである一方、“免疫沈降” タイプの実験では、支持担体に抗体を固定化し、抗体と相互作用した抗原を検出もしくは測定する。Peptide IP assay は、免疫沈降を模倣したフォーマットであるため、ChIP に使用する抗体の特異性評価に最適である。実際、Peptide IP assay において高い特異性を示した抗体が ChIP-seq においても高い性能で機能している(4)。
本稿では、Peptide IP assay を用いて抗ヒストン翻訳後修飾抗体の特異性を評価する手順を紹介する。本手法はヒストンペプチドのみならず、ビオチン化したタンパク質やペプチドを抗原として用いることも可能であり、様々な抗体の特異性の定量的評価にも応用できるため、汎用性の高い抗体の性能評価法となり得る。
装置・器具・試薬
- フローサイトメーター(各社)
- マイクロチューブ用ローテーター(各社)
- プレートシェイカー(各社)
- ボルテックスミキサー(各社)
- 磁気ビーズスタンド(各社)
- 96well プレート(各社)
- 96well フィルタープレート(MultiScreenHTS HV, Millipore)
- マルチスクリーンバキュームマニフォールド(Millipore)
- Protein A or Protein G Dynabeads(Thermo)
- ビオチン化ペプチド(各社。著者らは、HPLC により精製した純度95%以上のペプチドを使用している。ヒストンペプチド配列は表1を参照すること。Cys 残基を利用したビオチンの化学修飾法については、文献3を参照すること)
- 蛍光標識ストレプトアビジン(各社)
実験手順
I マグネティックビーズへの抗体の固定化
- ビーズの準備
- ビーズと抗体の混合及び洗浄
II 抗体–抗原反応
- ビオチン化ペプチドの準備
- 抗体固定化ビーズとペプチドの混合及び洗浄
- 検出試薬の添加と洗浄
III フローサイトメトリーによる蛍光の検出
- 測定の準備
- サンプルの測定
IV データ解析と平衡解離定数(KD)値の算出
実験の詳細
I マグネティックビーズへの抗体の固定化
Peptide IP assay では、抗体–抗原反応中に抗体がビーズから解離することを防ぐため、抗体はビーズに強固に固定化されている必要がある。IgG 結合タンパク質である Protein A 及び Protein G の抗体への親和性は、動物種とサブクラスによって異なるため、実験者は使用する抗体に高い親和性を示す IgG 結合タンパク質を選択しなくてはならない。例えばマウス由来の抗体には Protein G ビーズが適しており、ラビット由来の抗体には Protein A ビーズが適している。本稿では、Protein A ビーズを用いた実験例を記す(3,4)。
1) ビーズの準備
Protein A Dynabeads をボルテックスミキサーでよく懸濁し、10 μL のビーズを100 μL のPBST/BSA [PBS, 0.5% (W/V) BSA, 0.1% (V/V) Tween20] 溶液に加え、ピペッティングにより攪拌する。マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。この操作をもう一度繰り返す。
2) ビーズと抗体の混合及び洗浄
1 μg の抗体を100 μL の PBST/BSA 溶液に添加し、ピペッティングまたはタッピングにより攪拌混合することで抗体希釈液を作る。抗体希釈溶液をステップ1で調製したビーズに添加し、4℃にて1時間、ローテーターを用いて攪拌混合する。1ビーズあたりの抗体固定化量が不均一となるため、抗体原液をビーズ懸濁液に直接添加することは避ける。攪拌混合後、マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。300 μL の PBST/BSA 溶液でビーズを懸濁し、マグネティックスタンドを用いてビーズを吸着させ、上清を捨てる。この操作をもう一度繰り返し、最後に抗体固定化ビーズを100 μL の PBST/BSA 溶液に懸濁する。抗体固定化ビーズは、ヒストンペプチドと混合する直前に PBST/BSA 溶液にて40倍希釈する。
II 抗体–抗原反応
抗体を固定化したビーズとビオチン化ペプチドを混合し、抗体–抗原結合反応を行う。抗原結合反応が平衡に達した後、抗体に未結合のペプチドを洗浄し、蛍光標識ストレプトアビジンを加える。ビーズ上の抗体と相互作用しているビオチン化ペプチドに蛍光標識ストレプトアビジンが結合することで、フローサイトメトリーにより抗体–抗原相互作用に依存した蛍光を検出することが可能となる(図1)。
1) ビオチン化ペプチドの準備
各抗体–抗原反応において、抗体固定化ビーズと等量のビオチン化ペプチドを混合するため、実験者は測定したい濃度の2倍濃いビオチン化ペプチドを準備する。平衡解離定数 KD を算出するため結合曲線を描きたい場合、複数のデータポイントが必要となるため、実験者はあらかじめ連続希釈した溶液を作製しておく。ペプチド濃度の決定については、「工夫とコツ」を参照すること。希釈液は PBST/BSA である。
2) 抗体固定化ビーズとペプチドの混合及び洗浄
10 μL の希釈した抗体固定化ビーズと10 μL のビオチン化ペプチドを混合する。サンプル数が多い場合は、96well プレートを用いると便利である。抗体の親和性が著しく高く(例えば抗体の KD 値が picomolar 領域) 、抗原ペプチド濃度を低くする必要がある場合には、抗体固定化ビーズの量を減らす、もしくは反応体積を増やすことで ligand depletion(抗体量に対する抗原量の不足)を防ぐことができる。4℃で30分間振盪させた後、ビーズとペプチドの混合液を96well フィルタープレートに移す。バキュームマニフォールドを用いて溶液を吸引し、300 μL の PBST/BSA を各 well に添加し吸引することで、抗体固定化ビーズを洗浄する。洗浄操作を2回繰り返す。
3) 検出試薬の添加と洗浄
20 μL の10 μg/mL 蛍光標識ストレプトアビジン(著者らは、Streptavidin Dylight650(Thermo)を使用している)を各 well に添加する。4℃で30分間振盪させた後、バキュームマニフォールドを用いて溶液を吸引し、300 μL の PBST/BSA を各 well に添加し吸引することで、抗体固定化ビーズを洗浄する。洗浄操作を2回繰り返す。
III フローサイトメトリーによる蛍光の検出
1) 測定の準備
実験者はまず、ペプチドと反応させていない抗体固定化ビーズをフローサイトメーターに流し、scatter plot にてビーズ集団にゲートをかける。Dynabeads は粒径が均一であるため、scatter plot にてビーズ集団が集約しており、ビーズを判別しやすい。その他、検出感度を調整するためのゲイン値などの設定は、装置によって異なるため、実験者が最適な値を設定する。
2) サンプルの測定
100 μL の PBST/BSA 溶液を用いてビーズを懸濁し、フローサイトメーターに流すことで抗体に結合したペプチドに由来する蛍光を検出する。ゲート内のビーズカウントが、500以上あれば信頼度の高い平均蛍光強度値(MFI: Mean Fluorescence Intensity)を得ることができる。
IV データ解析と平衡解離定数(KD)値の算出
フローサイトメーターに付属のソフトウエア、もしくは FlowJo などの解析ソフトを用いて、各サンプルの MFI を算出する。抗体の標的ペプチドに対する結合曲線は、ペプチド濃度に対して MFI をプロットすることで描くことができる。平衡解離定数(KD 値)は、下記の式-1を用い、非線形最小二乗法にてカーブフィッティングを行うことで算出することができる。
\[\mathrm{MFI}(x) = \mathrm{MFI}(0) + \{\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\} \times (\frac{x}{K_D + x}) \tag{式-1}\]ここで \(x\) はペプチドの濃度、\(\mathrm{MFI}(x)\) はあるペプチド濃度での MFI、\(\mathrm{MFI}(0)\) はペプチドが非存在下(ペプチド濃度が0)での MFI 、\(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) は飽和濃度のペプチドを加えたときの \(\mathrm{MFI}\) から \(\mathrm{MFI}(0)\) を差し引いた値である。カーブフィッティングに際しては、KD 値のみを未知数として扱い、\(\mathrm{MFI}(0)\) 及び \(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) は固定値として扱う。非標的ペプチド(Off targets)への平衡解離定数は、\(\mathrm{MFI}(\infty) - \mathrm{MFI}(0)\) がすべての抗原ペプチドで等しくなると仮定してカーブフィッティングを行い、算出する。Peptide IP assay による抗体特異性評価の例として、抗ヒストン H3 トリメチル化 Lys9 抗体について結合評価を行った結果を図2に示す。
工夫とコツ
ビーズの取り扱いについて
ビーズは沈殿しやすく、いずれのステップにおいてもビーズをよく懸濁することが重要である。
ビーズの種類について
本稿では、マグネティックビーズを用いた Peptide IP assay を紹介したが、Protein A や Protein G がコーティングされているポリスチレンビーズを使用することも可能である(3,4)。ポリスチレンビーズを用いる場合、遠心によりビーズを分離する。
非特異吸着の確認
実験者は、抗体を固定化していないビーズとペプチドを混合し、ビーズが抗原ペプチドに非特異吸着しないかを確認する必要がある。著者らは、H4K20me1 ペプチドが Protein A をコーティングしてあるポリスチレンビーズ(Spherotech)に非特異的に吸着することを観測している(3)。
濃度が不明な市販抗体について
ポリクローナル抗体や血清において、抗体濃度が不明な場合がある。その場合、まず1 μL の抗体をビーズに固定化し、測定を行う。MFI が低く、結合曲線が描けない場合、a) ビーズへの抗体固定化量が少ない、b) 抗体自身の結合特性である、可能性がある。原因が前者である場合は、抗体量を増やすことで問題を解決できる。
抗原濃度域の決定について
抗体の標的抗原に対する見かけの平衡解離定数を算出したい場合、KD 値の濃度を含む抗原濃度域にて結合曲線を描く必要がある。そのため著者らは、まず標的抗原のみを用いて予備実験を行い、抗体の標的抗原に対する KD 値を見積もることを勧める。予備実験においては、数 μM の抗原を最大濃度として用い、濃度を細かく振る必要は無い。予備実験より得られた結合曲線からおおよその抗体の KD 値を見積もることができる。
本手法で算出される見かけの平衡解離定数(KD 値)について
Peptide IP assay においては、a) 抗体と Protein A/G との相互作用、b) 抗体と抗原の相互作用、c) ビオチン化ペプチドと蛍光標識ストレプトアビジンの相互作用、の3つの相互作用が存在する。IgG 結合タンパク質の抗体への結合親和性は、KD 値が~10-9 M と高く(7)、加えて avidity 効果(多価効果)により見かけの親和性はさらに高くなる。またストレプトアビジンのビオチンへの結合親和性は KD 値が~10-15 M と非常に高い。すなわち、(a) と (c) の相互作用は、(b) 抗体–抗原相互作用と比較して非常に強いため無視でき、本手法で算出される見かけの KD 値は (b) 抗体–抗原相互作用を反映していると著者らは考えている。
ビオチン化組換え抗体断片の利用
本稿では、IgG 抗体を用いた Peptide IP assay を紹介したが、ビオチン化した組換え抗体断片(Fab など)を用いることも可能である。その場合、ストレプトアビジンがビーズに固定化されている M280 Dynabeads(Thermo)を使用する。詳しくは、文献4を参照すること。
文献
- Bradbury, A. & Plückthun, A., Nature, 518, 27–9 (2015)
- Egelhofer, T.A. et al., Nat. Struct. Mol. Biol., 18, 91–3 (2011)
- Nishikori, S. et al., J. Mol. Biol., 424, 391–9 (2012)
- Hattori, T. et al., Nat. Methods, 10, 992–5 (2013)
- Rothbart, S.B. et al., Mol. Cell, 59, 502–11 (2015)
- Chao, G. et al., Nat. Protoc., 1, 755–68 (2006)
- Sidorin, E.V. & Solov’eva, T.F., Biochemistry (Moscow), 76, 295–308 (2011)
