

概要
現在、大規模なスクリーニング系により、膨大な量の蛋白質間相互作用に関するデータベースが構築されつつある。一方、これらの結合を定量的に解釈し、個々の反応の特異性を記述することは、重要であるにも関わらず、簡便に行うことは難しい。
蛋白質間相互作用の定量解析には、表面プラズモン共鳴法や滴定型熱量計が、最近の論文では良く見られるようになった。将来的には、これらの測定法が、一般的に誰でも利用することができるようになるかもしれないが、少なくとも、現時点で、いずれの装置も安価なものとは言いがたい。
ここでは、腸管出血性大腸菌 III 型分泌系に関与する蛋白質である EspB とヒト・αカテニン C 末ドメインの結合を例にして、蛍光偏光解消法を利用した比較的安価な蛋白質間相互作用解析法を示す。本法はサンプルの調製の段階で、若干、テクニックを要するが、解離・会合定数を比較的安価に見積もることができる点で優れている。
イントロダクション
蛍光偏光解消の原理については、多くの教科書に詳細が記述されている。本プロトコールは、原理を解説することが目的でないため、詳しい原理を勉強したい方は、他の文献(1-2)を参照していただきたい。そこで、以下には、実験を進めるために、最低限必要な概念を書くだけに止める。
フルオレセインなどの蛍光分子には、遷移モーメントが存在する。蛍光分子の遷移モーメントと一致する直線偏光により、蛍光分子を励起した場合、発せられる蛍光も遷移モーメントと同じ傾きの偏光を示す。一方、溶液中に漂う蛍光プローブはブラウン運動などにより、回転しているため、遷移モーメントも時々刻々と変化する。よって、もし、蛍光寿命が、分子の回転運動よりも、十分長ければ、励起光と異なる角度の偏光が、蛍光では発せられる。蛍光プローブの回転拡散係数が小さい場合、遷移モーメントが速く、且つ、分子ごとに異なる方向に時間とともに変化するので、蛍光は徐々に偏光性を失うことになる。つまり、正に『蛍光偏光が解消する』。蛍光偏光解消では、このような解消にかかる時間スケールの変化を観察する方法もあるが、今回のように滴定実験を行う場合には、導入として、静的な測定の方が簡便である。すなわち、大きさの異なる分子に、励起光を当て続けたときの蛍光偏光の平均的な傾きを観察する方法をここでは考える。
もし、特定の蛋白質に蛍光プローブを共有結合で導入し、その蛋白質が他の蛋白質と結合し、単量体から、ヘテロ二量体になった場合、分子量が増大するため、偏光の傾き具合が、小さくなる。この現象を利用し、滴定曲線を得ることで、蛋白質間の結合をモニターし、さらに、そこから結合(解離)定数を非線形のカーブフィッティングなどで、解析することが可能となる。
偏光の傾き具合を表す指標には、異方性(anisotropy: \(r\))があり、その値は、以下の式で表される。
\[ r = ( I_{\mathrm{vv}} - G{\ }I_{\mathrm{vh}} ) / (I_{\mathrm{vv}} + 2{\ }G{\ }I_{\mathrm{vh}}) \tag*{式(1)} \]
\[ G = I_{\mathrm{hv}} / I_{\mathrm{hh}} \tag{式2} \]
ここで、\(I_{\mathrm{vv}}\), \(I_{\mathrm{vh}}\), \(I_{\mathrm{hv}}\), \(I_{\mathrm{hh}}\) は、異なる偏光度を持った励起光、蛍光での蛍光強度を表し、添字の \(\mathrm{v}\), \(\mathrm{h}\) は偏光の角度を表す。すなわち、\(\mathrm{v}\) は水平(vertical)、\(\mathrm{h}\) は垂直(horizontal)方向の偏光を示す。各添字の内、先に書かれたものが、励起側の偏光の角度を示し、後に書かれたものは、蛍光側の角度を示す。つまり、例えば、\(I_{\mathrm{vh}}\) とは、水平の偏光で励起した時の垂直方向の偏光成分の示す蛍光強度を表す。なお、\(G\) は装置固有の補正値である(詳細は教科書を参照)。
本プロトコールは、上記の現象を利用し、二分子間の結合過程を滴定実験により追跡し、その結果を非線形カーブフィッティングで解析するまでの一連の仕事について、EspB とαカテニン C 末端ドメインを例にして(浜口ら未発表データ)、記述する。
装置・器具・試薬
- 蛍光分光装置(各社)
- 直線偏光子、又は偏光フィルム(各社)
- 蛍光分光用セル(各社)
- 蛍光ラベル化剤(フルオロセインイソチオシアナート、5-ヨードアセトアミドフルオレセインなど)
- αカテニン C 末端ドメイン(分子量:25.9 kDa、235残基)
- EspB(分子量:32.6 kDa、313残基)
※使用する蛋白質については、既に精製品が存在していることが前提である。蛋白質の精製については、他のプロトコール集参照。
実験手順
- 蛋白質への蛍光ラベル(5-ヨードアセトアミドフルオレセイン: 5-IAF)の導入
- ベル化効率、蛋白質濃度の決定
- 測定サンプルの調製
- 測定
- 解析
実験の詳細
第1日
1. 蛍光ラベル(5-ヨードアセトアミドフルオレセイン: 5-IAF)の EspB への導入
EspB には46番目に遊離のシステイン残基が存在する。このシステインに選択的に 5-IAF を導入する方法を以下に示す。
まず、1 mg/ml(~ 30 μM)EspB、10 mM トリス-塩酸(pH 8.0)の溶液を 1 mL、調製する。この際、特にチオール基を持つ還元剤(ジチオスレイトールやβ-メルカプトエタノール)の混入が心配される場合には、透析や脱塩カラムなどであらかじめ処理する。
次に、10 mM の 5-IAF、10 mM トリス-塩酸(pH 8.0)、15 μL を上記の蛋白質溶液に添加する。これで、約150 μM の5-IAF が添加されたことになる。つまりチオール基の5倍のモル量の蛍光試薬が加えられたことになる。
蛋白質溶液を室温、暗所で1-2時間保温した後、10 mM リン酸ナトリウム緩衝液(pH 7.0)に対して、透析し、余分な未反応の 5-IAF を取り除く。なお、以下の測定を急ぐ時は、脱塩カラムやより高精度のゲル濾過カラム、HPLC などを使用する。
第2日
2. ラベル化効率、蛋白質濃度の決定
上記の方法で、5-IAF により蛍光ラベルした EspB(IAF-EspB)のラベル化効率は分光学的に決定する。溶液内の蛋白質濃度と、導入された 5-IAF の濃度を見積もり、その総対比でラベル化効率を判断する。ここでは分光光度計を使った簡易法を記述する。なお、詳細は、工夫とコツ「蛍光ラベル化された蛋白質の濃度」を参照していただきたい。
5-IAF は491 nm に吸収極大をもち、その分子吸光係数(\(\varepsilon_{\mathrm{IAF,491}}\))は \(82,000{\ }\mathrm{M}^{-1}\) である。これを用いて、導入された 5-IAF の濃度(\(c_{\mathrm{dye}}\))を定量する。一方、5-IAF は 280 nm 付近の光も吸収する。通常、pH 7.0 付近では、280 nm での 5-IAF の吸収は 491 nm の吸収の30%である。よって、蛋白質濃度(\(c_{\mathrm{prot}}\))は、蛋白質の 280 nm での分子吸光係数を \(\varepsilon_{\mathrm{prot}, 280}\) とすると以下の式で表される。
\[ c_{\mathrm{prot}} = (A_{280} - A_{491} \times 0.3) / \varepsilon_{\mathrm{prot}, 280} \]
なお、ここで \(A_{280}\),\(A_{491}\) は、それぞれ 280 nm、491 nm での溶液の吸光度とする。このような測定により、修飾効率:\(F\) は、\(F = c_{\mathrm{dye}} / c_{\mathrm{prot}} \times 100\)(%)が見積もられる。
実際に、以下の実験で用いた IAF-EspB の修飾効率は、99%であった。
原理上、修飾率が低い場合でも、下記の実験を進めることができる(工夫とコツ「ラベル導入と測定に関する注意事項」参照)。なお、この分光測定は、IAF-EspB の蛋白質濃度を決定する上でも重要であるので、必ずやらなければならない。
3. 測定サンプルの調製
IAF-EspB 濃度 0.05 μM(~0.001 mg/ml)で、異なる濃度のα-カテニン C 末端ドメインを含む溶液を数本、調製した。実際の手順は以下の通りである。
まず、蛋白質の濃度決定をした際に、上記 1. の手順で調製した IAF-EspB の濃度が 10 μM であり、α-カテニン C 末端ドメインのストック溶液の濃度を、54 μM(~1.4 mg/ml)とした。筆者の使用する蛍光セルは、一回の測定で 300 μL 必要なので、表1に記載した量の溶液を混合し、サンプルを調製した。なお、いずれの蛋白質も 10 mM リン酸ナトリウム緩衝液に溶かしたものを使用している。
これらの溶液を調製後、恒温槽にて10℃(実際に測定したい温度)で一晩保温し、翌日の測定に用いる。
第3日
4. 測定
蛍光光度計は、励起光 419 nm で励起し、520 nm の蛍光を観察するように測定条件を設定し、時間変化測定を行う。\(I_{\mathrm{vv}}\) を測定できるように、偏光子を励起側、蛍光側にそれぞれ取り付ける。上記 3. で用意したサンプルを蛍光用セルに移し、蛍光光度計にセットし、蓋をする。蛍光は温度に依存するので、セルの温度を一定に保つように、循環水をホルダー内に回すなど工夫する。セル内の温度が一定になるまで、少なくとも5分は待った方が良い。なお、反応系の平衡がずれるので、急激な温度変動も避けるべきである。ここでの実験は10℃で行ったもので、前日からの保温温度と一致させるのが、時間短縮のためのコツである。
測定を開始し、シグナルが一定になったと思うところで、励起側の偏光子を90度傾け、\(I_{\mathrm{vh}}\) のシグナルを測定し、シグナルの変動を記録する。同じ手順で \(I_{\mathrm{hh}}\)、\(I_{\mathrm{hv}}\) の順番に測定する。これを各サンプルについて行いそれぞれの結果をファイルに保存し、次の解析に用いる。
5. 解析
解析には、さまざまな表計算ソフトを使用することが可能であるので、ここでは、どのソフトウェアでも共通と思われる事項のみを記載する。
上記 4. で測定したデータをそれぞれ表計算ソフトに取り込む。それから、蛍光強度を測定時間に対してプロットする。\(I_{\mathrm{vv}}\)、\(I_{\mathrm{vh}}\)、\(I_{\mathrm{hh}}\)、\(I_{\mathrm{hv}}\) に相当する領域のデータの平均値をそれぞれのソフトウェアで推奨の方法で計算し、各α-カテニン C 末ドメインの濃度ごとに整理する。その後、各濃度に対する \(r\) を計算し、その値をα-カテニン C 末ドメイン濃度に対して、プロットする。ここで、特に問題なければ、図1に示すような飽和曲線が得られる。
これを各反応系にあった反応モデルに従って、非線形の最小自乗法によりカーブフィッティングし、必要な解離・会合のパラメータを得る。ちなみに、ここで示した例では、最も簡単な \(1 : 1\) の解離会合系のモデルで解析することができた。
添加した IAF-EspB の濃度を \(c_{\mathrm{E}}\)、α-カテニン C 末ドメインの濃度を \(c_{\alpha}\) とし、両者の解離定数を \(K_{\mathrm{d}}\) とすると、会合状態の濃度:\(c_{\mathrm{E}\alpha}\) は、以下の式で表される。
\[ c_{\mathrm{E}\alpha} = \frac{( c_\mathrm{E} + c_\alpha + K_\mathrm{d} ) − \sqrt{( c_\mathrm{E} + c_\alpha + K_\mathrm{d} )^2 − 4c_{\mathrm{E}}c_{\alpha}}}{2} \tag*{式(3)} \]
蛍光偏光度は、解離状態、会合状態の IAF-EspB の濃度比率(\(c_{\mathrm{E}\alpha} / c_{\mathrm{E}}\)、\((c_{\mathrm{E}} - c_{\mathrm{E}\alpha}) / c_{\mathrm{E}}\))によって決まるので、それぞれの状態が100%存在する時の蛍光偏光度を、それぞれ \(r_{\mathrm{E}\alpha}\)、\(r_{\mathrm{E}}\) とすると、各条件での蛍光偏光度 \(r\) は、
\[ r = r_{\mathrm{E}} ( c_\mathrm{E} - c_{\mathrm{E}\alpha} ) / c_{\mathrm{E}} + r_{\mathrm{E}\alpha} c_{\mathrm{E}\alpha} / c_{\mathrm{E}} \tag*{式(4)} \]
で表される。ここでは、式(4)に基づき、図1のデータをカーブフィッティングすることで、解離定数 \(1.5 \times 10^{-6}{\ }\mathrm{M}\) を得た。
工夫とコツ
蛍光ラベルの選択
蛍光ラベルの導入方法には、大まかに分けて、
- 遊離のシステイン側鎖のチオール基を介した修飾方法、
- リジン側鎖に存在するε-アミノ基や N 末端のα-アミノ基を介した修飾方法
の二種類がある。
前者 a は「遊離のシステイン残基が配列内に存在し、蛍光修飾により蛋白質の構造や安定性に影響をあまり与えない」ことが必要不可欠な条件である。最近では、リコンビナント蛋白質の合成手法により、本来存在しない遊離のシステイン残基を蛋白質の末端に導入することで、比較的容易にこれら条件を与えることが可能になってきた。一方、このような末端の改変が蛋白質の性質に影響を与える可能性もあるので、注意が必要である。使用することのできる蛍光色素側の官能基は、ヨードアセトアミド基やマレイミド基などがある。チオール基修飾性であるために、溶媒に DTT やβ-メルカプトエタノールなどのチオール基を含む還元剤を使用することができない。
後者 b の場合、リジン側鎖もしくはブロックされていない N 末端α-アミノ基があれば良いので、ほとんどの蛋白質について実施可能である。逆に、あるアミノ基に選択的に修飾色素を導入することは容易ではない。
通常、N 末端α-アミノ基の方が、リジン側鎖のε-アミノ基よりも、解離の pKa が低く、低 pH(~7程度)でも修飾反応が進行する。従って、pH 7.0、室温以下で反応させると、それなりの選択性を持って N 末端α-アミノ基に蛍光色素を導入することができる。
使用できる蛍光色素側の官能基は、イソチオシアナート基やスクシンイミジル基などがある。これらの官能基はアミノ基修飾性であるため緩衝液にはトリスなどのアミン系試薬を使うことができない。よって、通常、リン酸緩衝液が推奨される。
なお、通常、b で使用する蛍光試薬の方が、a で使用できるものよりも安価であることも、修飾剤の選択において重要な要素である。
蛍光ラベル化された蛋白質の濃度
通常ならば、ラベル化効率を測定する際に測る 280 nm の吸収により、蛋白質濃度を決定する。しかし、運悪くトリプトファン残基、チロシン残基が配列に存在せず、280 nm の特徴的な吸収を示さない蛋白質の場合、他の方法を使用しなければならない。また、修飾により、分子吸光定数が変化する場合などもあり得るので、本質的に芳香族側鎖の吸収を用いるべきかどうかという疑問も残る。
別法として、BCA 法、ブラッドフォード法、ローリー法などのプロテインアッセイ法を用いる場合、蛍光ラベルの寄与をしっかりと見積もる必要がある。使用する分光光度計にも依存するであろうが、蛍光プローブと芳香族が 280 nm 付近に示す吸収は、通常互いに非常に大きく、しばしば大きな測定誤差を伴う。そこで、芳香族の有無に関わらず、BCA 法を用いる場合が、多くの文献で見受けられる。いずれにせよ、興味ある方(あるいは不安な方)は、一度、BCA 法と 280 nm の吸収を用いて得られる結果を比較してみられることをお薦めする。しかし、両者の結果に大きな隔たりがあった場合、どちらが正しいのか迷わないためにも、BCA 法に伴う測定誤差(かなりの点で実験者に依存する)も、それなりに認知しておかなければならない。あるいは、アミノ酸分析など、些か古典的な手法を使用して、濃度決定することもできる。
もし、今、蛍光を入れることを考えている蛋白質に芳香族環がなく、濃度決定が煩雑であると予想される場合、別の考え方は、ラベルを入れる蛋白質を変えることである。蛋白質間相互作用の系で、両蛋白質ともにトリプトファン残基、チロシン残基が配列に存在しないことは、非常に稀であろう。このような場合、280 nm の測定で濃度決定することができる蛋白質の方に蛍光色素を導入する方が、無難である。しかし、変異体解析などによる結合サイトの同定など、派生的な実験を行う場合、280 nm の吸収を示さない蛋白質をラベルした方が、その後の解析に都合が良いことも想定できる。個人的には、こういう場合は、リコンビナント系を用いて、蛋白質末端など反応に当たり障りのないと想定できる箇所にトリプトファン残基を加えた蛋白質を作る方が、良いと思われる。
ラベル導入と測定に関する注意事項
蛍光偏光解消により、分子間相互作用を解析する場合、「蛍光ラベルを導入した際に、結合反応の親和性に影響していないか?」、「滴定の過程で蛍光プローブの蛍光寿命(強度)に変化がないか?」ということ考慮しなければならない。これら両者の問題は、それなりに関連しており、プローブの蛍光寿命(強度)に変化があるということは、その領域が結合面に関与している可能性を示唆しており、そのような条件では、結合親和性に影響が少なからずあると予想できる。
つまり、蛍光偏光解消実験では、少なくとも反応親和性に影響を与えると考えられる部位に蛍光ラベルを導入することは避けるべきである。しかし、その部位の選択は非常に難しい。多くの場合、N 末端、C 末端が導入箇所の候補となるが、最低限、プローブの蛍光強度に変化がないことを確認するべきである。また、ラベル導入箇所による結合親和性における影響を見積もるためには、ラベル化効率の異なる蛋白質を用いて滴定実験を行い、得られる結果が大きく異ならないことを確認することもできる。一番確実なのは、分子表面の様々な部位にシステイン残基を導入した変異体をいくつか作り、蛍光プローブ導入に適切な場所をマッピングするのが最良である(4)。この方法は、手間がかかるが、同時に、蛋白質の結合面の探索にも知見を与える点で、有用である。最終的には、表面プラズモン共鳴や滴定型熱量計を用いて、同様な滴定実験を行い、蛍光偏光解消の結果と比較することができれば、より確実であろう。
チオール基への修飾
遊離のチオール基(システイン)を含む蛋白質では、長時間保存している間に分子間で架橋されてしまっていることもある。もし、修飾率が低い場合は、この可能性を、まず疑う。対処法としては、一旦、ジチオスレイトールなどで還元しなおし、脱塩カラムで余分な還元試薬を取り除いた蛋白質を修飾反応に使用する。なお、蛋白質内にジスルフィド結合を持つ蛋白質については、還元剤の量と反応時間を調整して、分子間架橋のみを還元するように工夫する必要がある。
蛋白質濃度の決定法
蛋白質の濃度は、分光学的に決定することをお勧めする。なお、蛋白質の分子吸光係数は、内在のトリプトファン側鎖、システイン側鎖、チロシン側鎖の数から、概算することができる。筆者は専ら ExPASy(kr.expasy.org)上の ProtParam tool を愛用している。
もし、不幸にして、使用している蛋白質に上記のアミノ酸がない場合は、プロテインアッセイキットを用いるのが簡便である。
非修飾蛋白質の測定濃度範囲
通常の滴定実験を行う準備にとりかかる際、もしも結合・解離定数の値が大体予測できていれば、どの程度、相手方蛋白質の量を振れば良いか、表計算ソフトなどで、シミュレーションすることができる。一方、良くわからない場合は、最初に、0,0.1,1.0,10 μM など、相手方蛋白質の濃度を、一桁ごとに違えたサンプルを調製し、予備実験をすれば良い。おそらく、この手の実験をするに当たって、プルダウンアッセイなどの実験が既に行われており、対象蛋白質間の相互作用があることは確認されていると思われる。プルダウンにより、どの程度の強さの分子間相互作用が解析できるのかは、筆者自身不明であるが、実験スケールで用いる蛋白質濃度は、せいぜい 100 μM 以下であろうと予想される。よって、そのような条件で確認できる結合反応に対する解離定数は \(1 \times 10^{-3}{\ }\mathrm{M}\) を超えることはないと思われる。よって個人的には、相手方蛋白質の濃度は 100 μM 以下で十分であると考えている。逆に言えば、それ以上に蛋白質濃度を上げる必要があるなら、その反応の特異性も低い可能性も疑うべきかもしれない。
偏光子と蛍光光度計
本実験で最も重要なのは励起、蛍光の両方で利用する偏光子(偏光板)と蛍光光度計である。偏光子については、各社、それぞれの分光器に対応した純正品を市販している。純正品の場合、分光器への取り付けが、前もって想定されているため、比較的容易にセッティングすることができ、確実に測定できるという安心感がある。しかしながら、通常、少し高額である。比較的頻繁に測定を行う方や予算に余裕がある方なら、ソフトウェアと連動して、一回のボダン操作で自動的に偏光子を回転させ、全てのデータを取り込んでくれるオプションや、さらに蛍光プレートリーダータイプのハイスループット型まである。一方、最も安価なものは、液晶ディスプレー部品やカメラの偏光用として市販されている偏光子を普段、使っている蛍光光度計に取り付ける方法もある。他の偏光子の候補には、グラムトンプソンプリズムなどが考えられるが、若干、高価で且つ、比較的大きいので、蛍光光度計の試料室内に収まるサイズを検討しなければならない。
筆者は、日本分光社製 FP-6500 蛍光分光計を愛用しているが、この装置には励起側、蛍光側にそれぞれ、四角いフィルターを取り付ける溝がある。そこで、ポラロイド社製の HN32直線偏光フィルターを正四角形に切ったものを、分光器の溝に取り付け使っている。このフィルターは、筆者の共同研究者である、京都工芸繊維大・田中直毅 先生に教えていただいた。これだと、投資額数千円ですむ。しかしながら、残念ながら、数年前にポラロイド社が、セルロイド製の偏光子の製造をストップしているようで、国内で手に入れることは困難なようだ。ちなみにポラロイドとは、偏光子(polarizer)とセルロイド(celluroid)をくっつけた造語だそうです。
解析ソフト
多くの表計算ソフトウェアを使って、カーブフィッティングを行うことができるので、それぞれの実験者が慣れ親しんだソフトを使って解析するのが一番であると考えられる。なお、筆者及び筆者の関係者は、この手のカーブフィッティングでは、IGOR PRO(Wavemetrics Inc., USA)を使用してきた。本実験に限らず、多くの場合、カーブフィッティング解析のためには、反応モデルに対応する理論式を書く必要がある。このソフトウェアは、ユーザー側で理論式を自由に書き換えられる他、他の機能も充実しており、筆者の執筆する論文でも、度々登場するソフトウェアの一つである。もし、非線形のカーブフィッティングを行うことができるソフトウェアが入手できない場合で、且つ、1:1 の結合系である場合、Scatchard プロット(結合した分子の量/遊離の分子の量 vs 結合した分子の量)により、解離定数を解析することも可能である(3)。
文献
- 木下一彦、御橋廣眞 編 螢光測定 - 生物科学への応用(日本分光学測定法シリーズ(3))学術出版センター、1983年
- Lakowicz, J. R. Principles of Fluorescence Spectroscopy 3rd Ed. Springer, 2006.
- Bergethon, P. R.(谷村吉隆ら訳)ベルゲゾン生化学の物理的基礎、シュプリンガー・フェアラーク東京、2004年
- Tanaka, N. & Fersht, A. R. Identification of substrate binding site of GroEL Minichaperone in Solution. J. Mol. Biol. 292, 173–180 (1999)
-
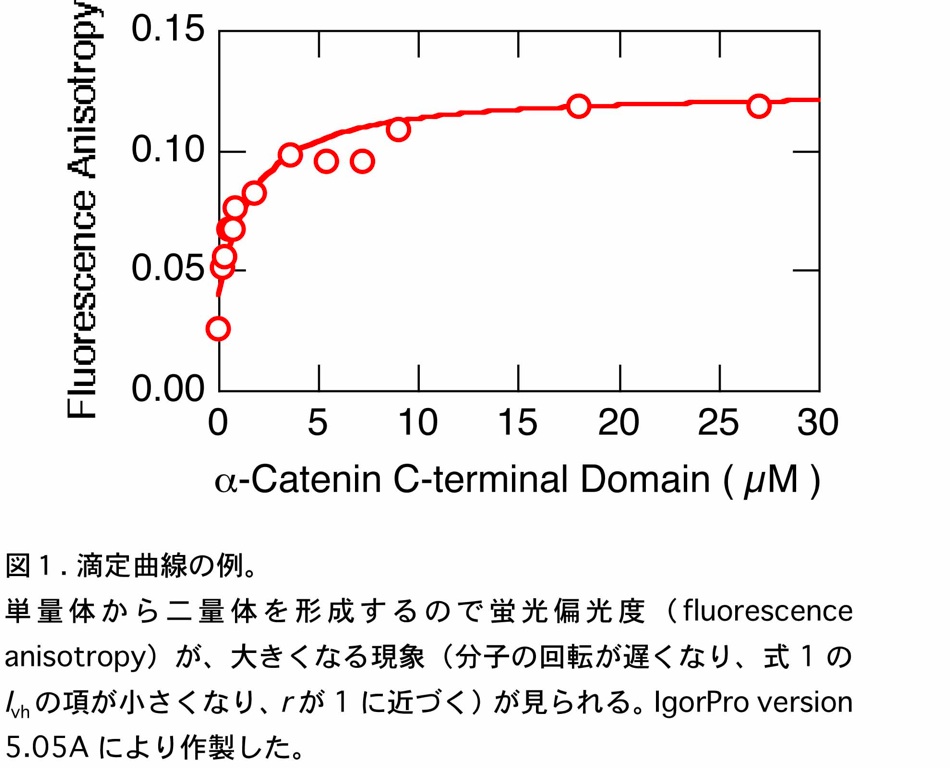
図1: -
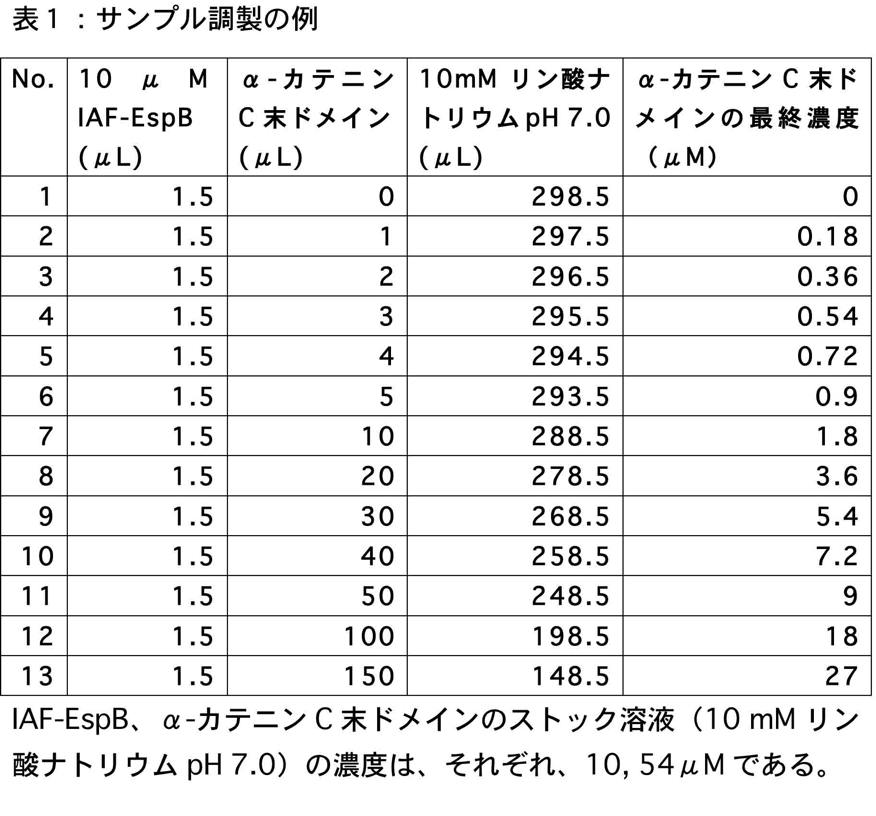
表1:
