

全体の概要
発現ベクターを構築する上での、DNAに優しい切出し、形質転換の効率向上、時間短縮、コストダウンに役立つ DNA の泳動方法やライゲーション方法の情報提供。
主な内容
- UV 照射をしないゲルからの DNA 切出し
- 高効率コンピテントセルの自作方法
- 挿入断片が入ったコロニーの経済的な目星の付け方
1. UV照射をしないゲルからのDNA切出し
装置・器具・試薬
- 核酸泳動用のアガロースゲル(1%前後の固さ)と電気泳動槽(Mupid など)
- CLEAR STAIN Blue(ニッポンジーン社製)
- Quick Ligation™ Kit(New England Biolabs 社製)やIn-Fusion® HD Cloning Plus(ClonTech/タカラバイオ社製)
制限酵素処理後の DNA は、アガロースゲル電気泳動で目的断片を他の断片から分離する。通常は、泳動後にエチジウムブロマイド(EtBr)で染色し、暗室の UV イルミネーター上でバンドを確認することが広く行われている。しかし、UV 照射による DNA の損傷のために、変異頻度の上昇やライゲーション効率の低下が問題となる場合がある。初心者が作業に馴れずにもたついて長時間 UV をゲルに照射してしまう際や、UV イルミネーターを新品に交換した際に、実験がうまくいかなくなる原因の一つとして過剰な UV 照射による DNA の損傷が考えられる。また、EtBr の代替品となる染色試薬も市販されているが、EtBr に比べると高価だったり専用の検出装置が必要とされるケースが多い。
筆者らは UV 照射をせずにゲルからの切り出しを行うために、ニッポンジーン社から販売されている CLEAR STAIN Blue を利用している。本品はアガロースゲルと DNA を試薬で染色して、蒸留水で脱色することで DNA 断片が可視化される (1)。そのため、わざわざ暗室と実験室を往復することなく、スムーズに DNA の精製ステップに進むことができる。
切出し後のライゲーションについても、16℃などの低温インキュベータを必要とせずに、室温で短時間に実施できる酵素が販売されている。New England Biolabs(NEB)社の Quick Ligation™ Kit (2)やプロメガ社の LigaFast™ Rapid DNA Ligation System (3) は室温5分間の反応でライゲーション反応を完了できる。これらは kit や system と名が付いてはいるが、実際には DNA 溶液にバッファーと酵素を加えるだけである。付け加えておくと、NEB 社の T4 DNA ligase は実は室温で利用できるので、若干反応時間が長くはなるがよりコストを圧縮することが可能である (4)。また、PCR 断片などをクローニングする際は、タカラバイオ社から販売されている In fusion cloning が便利である (5)。このキットでは、切断したベクターの末端配列15塩基を目的 DNA 増幅用プライマーに付加して PCR 産物を調製するだけで、比較的大きな断片や複数断片の同時クローニングが格段に高い効率で可能である。ベクターと PCR 断片に1/5量の試薬を加え、50℃で15分間反応させるだけでコンピテントセルへの形質転換が可能である。PCR 断片を制限酵素処理しなくてよいので、クローニングの自由度が高いのが大きな利点である。
CLEAR STAIN Blue を利用した実験手順
- 1%アガロースゲルでサンプルを泳動後、10倍希釈した CLEAR STAIN Blue 溶液にゲルを浸し、10分間緩やかに攪拌。
- ゲルを蒸留水で洗浄、10分間緩やかに攪拌。DNA 濃度が高い場合には洗浄直後にバンドが確認できる。
- 目的のバンドを切り出し・精製を行う(日本ジェネティクス社で販売している FastGene Gel/PCR Extraction Kit などを利用)。
New England Biolabs(NEB)社Quick Ligation™ Kit を利用した実験手順
- ライゲーションを行う DNA のサンプルを 10 μl の滅菌水に溶解する。
- 2X Quick Ligase Buffer を 10 μl 加え、よく溶解する。
- 1 μl の Quick T4 DNA Ligase を加え、25℃で5分間反応させる。その後、コンピンテントセルを使った形質転換を行う。
タカラバイオ社 In-Fusion® HD Cloning Plus を利用した実験手順
- ライゲーションを行う DNA のサンプルを 4 μl の滅菌水に溶解する。
- 5X In-fusion Enzyme Premix を 1 μl 加え、50℃で15分間反応させる。その後、コンピンテントセルを使った形質転換を行う。
2. 高効率コンピテントセルの自作法
市販のコンピテントセルを利用するのは経済的に負担が大きいので、超低温フリーザーでの保存も可能なコンピテントセルを自前で作成する。Inoue et al. (1990) の方法が多くのラボで少しずつ改変されて使われている (6)。作業時間は約半日、培養を除けば1時間少々。
装置・器具・試薬
- 大腸菌(各自の好みの菌株)
- Transformation Buffer(TB):950 mL の蒸留水に 10 PIPES(3.0 g)、15 mM \(\ce{CaCl2 \cdot 2H2O}\)(2.2 g)、250 mM \(\ce{KCl}\)(18.6 g)を溶解し \(\ce{KOH}\) で pH6.7 に調整する。さらに \(\ce{MnCl2 \cdot 4H2O}\) を 10.9 g(55 mM)加え、蒸留水で終量 1 liter に fill up。濾過滅菌後4℃にて保存。使用前に氷冷しておく。
- SOB 培地:Bacto Tryptone 20 g、Yeast Extract 5 g、5M \(\ce{NaCl}\) 2 mL、2 M \(\ce{KCl}\) 1.25 mL を終量 990 mL の水に溶解してオートクレーブ。60℃以下に冷めてから濾過滅菌した2 M Mg solution(1 M \(\ce{MgSO4 \cdot 7H2O}\) + 1 M \(\ce{MgCl2 \cdot 6H2O}\))を 10 mL 加える。冷蔵保存し、無菌的に利用する。
- 液体窒素、氷
- 超低温フリーザー(−80℃以下で使用)
- 50 mL サイズの滅菌済遠心管(ファルコンチューブ)、滅菌済サンプルチューブ、低温遠心機
実験手順
- 300 mL 三角フラスコで 50 mL SOB 培地で \(\mathrm{OD_{600}} = 0.5\) まで大腸菌を培養(低温で培養するほうが効率がよい、筆者らは25℃でオーバーナイトの培養をして、翌朝コンピテントセルを作成している)。
- 無菌的操作で培養液をファルコンチューブに移して10分間氷冷。
- 0–4℃で冷却しながら集菌(3000rpm で10分間)、上清は捨てる。氷冷した TB 16 mL を加え懸濁、10分間氷冷。
- 4℃で冷却しながら集菌(3000rpm で10分間)、上清は捨てる。
- 2 mL の氷冷したTBを加え懸濁、さらに150 μlのDMSOを加え(final 7%)、10分間氷冷。
- 100 μl ずつエッペンチューブに分注し液体窒素で急冷、その後−80℃で保存(1~2ヶ月は充分使用可能)。
利用法
市販のコンピテントセル同様の使用法で形質転換する。
- 氷上でコンピテントセルを溶解し、DNA sample を加え1時間氷冷(15–30分でも十分)。
- 42℃でヒートショックを30秒(あるいは120秒)与え、ただちに氷冷。
- 氷冷を2分した後、SOB 培地あるいは LB 培地 2 mL に懸濁、37℃で穏やかに1時間振盪する(振盪無しでも可、10~30分でも十分)。
- 集菌し plating、37℃で12~18時間培養。
3. 挿入断片が入ったコロニーの経済的な目星の付け方
発現ベクター構築の際に、形質転換後の多数の大腸菌コロニーの中から挿入断片が入ったコロニーを経済的かつ簡便に見つけだす(目星をつける)方法。Blue/white の判別ができない場合、挿入断片が保持されているかどうかをコロニー PCR や mini prep と制限酵素で確認したりするのは費用も手間もかかって大きな負担となる。そこで、まず本法で目星をつけておいて、目的の挿入断片が入っていそうなもののみ mini prep で精製して確認をすれば多少経済的・効率的になる。培養時間を除くと作業時間は約1時間。
装置・器具・試薬
- 滅菌した爪楊枝。順向き・逆向き二種類の方向で爪楊枝を 100 mL ビーカーに入れてアルミホイルでふたをしてオートクレーブしておく。使用後もオートクレーブによって再利用可。
- 大腸菌培養用のプレート(プラスミドを保持するための抗生物質等を含む)
- Cracking solution(3% w/v SDS、50 mM Tris-base、pH12.6)
- 核酸泳動用のアガロースゲル(1%前後の固さ)と電気泳動槽(Mupid など)
- 65℃のヒートブロックあるいはインキュベーター
- 泳動用色素(たとえば 100 mM EDTA、30% Sucrose、10% glycerol、10% Xylene cyanol、10% Bromo Phenol Blue の混合液、その他、タカラバイオや TOYOBO の制限酵素にオマケでついてくるものでも可能)
- 1.5 mL サンプルチューブ
- PCI(phenol : chloroform : isoamylalcohol = 25 : 24 : 1)
実験手順
- 形質転換で出たコロニーは滅菌した爪楊枝(順向き)を使い、一枚のシャ-レ当たり16個の plasmid を広げておく。コントロールとして空のベクタ-を保持した大腸菌も広げ、37℃で7–8時間培養(図1)。
- 1.5 mL サンプルチューブに Cracking solution を 50 μl ずつ分注する。
- つまようじの頭で菌体を採取し、Cracking solution に懸濁する(図2の白い部分が菌体)。懸濁すると粘りが出て糸を引くようになる。
- ふたをして65℃で10分間インキュベートする。
- ほぼ等量の PCI と数 μl の泳動用色素を加え5–10秒 Vortex(さらに時間を短縮したい場合は PCI と泳動色素を前もって混ぜてから分注する)。
- 13000rpm 程度で3~5分遠心。
- 上清を 20 μl 取りアガロース電気泳動、染色、観察(図3)。
- コントロールよりも上にバンドがくれば何かが入っている可能性大なので mini prep などで精製して確認する。
工夫とコツ
泳動は 150 V で30分ほど、Xylene cyanol の黄色のバンドがゲルの先端に届くぐらいまで流すと差がわかりやすい。
文献
- https://www.nippongene.com/siyaku/product/electrophoresis/pdf/bw149_clear-stain-blue.pdf
- http://www.neb.com/nebecomm/products_Intl/productM2200.asp
- http://www.promega.co.jp/Cre_Html.php?pGMPID=0206001
- http://www.neb.com/nebecomm/products_Intl/productM0202.asp
- https://catalog.takara-bio.co.jp/product/list.php?catcd=B1000345&subcatcd=B1000586#B1000586
- Inoue H., et al., Gene, 96, 23–28 (1990)
改訂履歴
2021年1月26日 改訂
- タイトルを「発現ベクターの構築に関わる Tips」から「プラスミド構築に関わる Tips」に変更。
- 「詳細」に記載の各項目の「概要」を削除。
- 項目1の項目名を「UV 照射をしないゲルからの DNA 切出し」に変更、「装置・器具・試薬」に記載の「GelMate2000」を削除(本文中においても削除)、「Gel Indicator™(BioDynamics Laboratory 社製)」を「CLEAR STAIN Blue (ニッポンジーン社製)」に変更(本文中の説明文も変更)、「In-Fusion® HD Cloning Plus(ClonTech/タカラバイオ社製)」を挿入(本文中に説明文を挿入)、本文に記載の「アルカリフォスファターゼ処理」と「EtBr を含んだゲル」の内容を削除、EtBr の代替品について言及。
- 項目2の「装置・器具・試薬」に記載の電気泳動の記述を削除、「実験手順」に記載の見出し番号を整理。
- 項目3の本文に記載の「TaKaRa」を「タカラバイオ」に変更、実験手順として見出し番号を挿入。
- 図1を削除、図番号を更新。
- 文献1を変更し、新たな文献5を追加し、文献番号を更新。
-
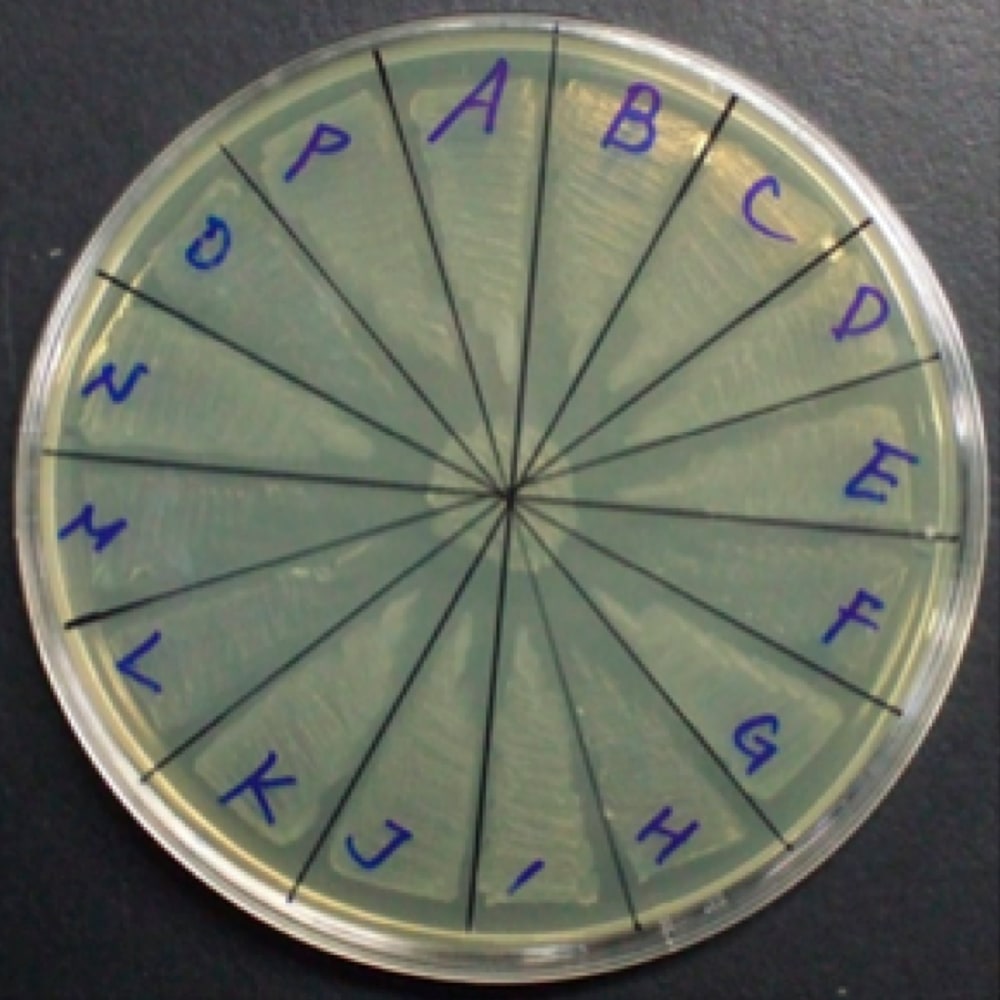
図1:コロニーをシャーレ16分割に広げる。中央にはコントロールとして空ベクターを持つ大腸菌を広げておく。 -

図2:爪楊枝の頭で菌体をかき取り cracking solution に懸濁する。 -
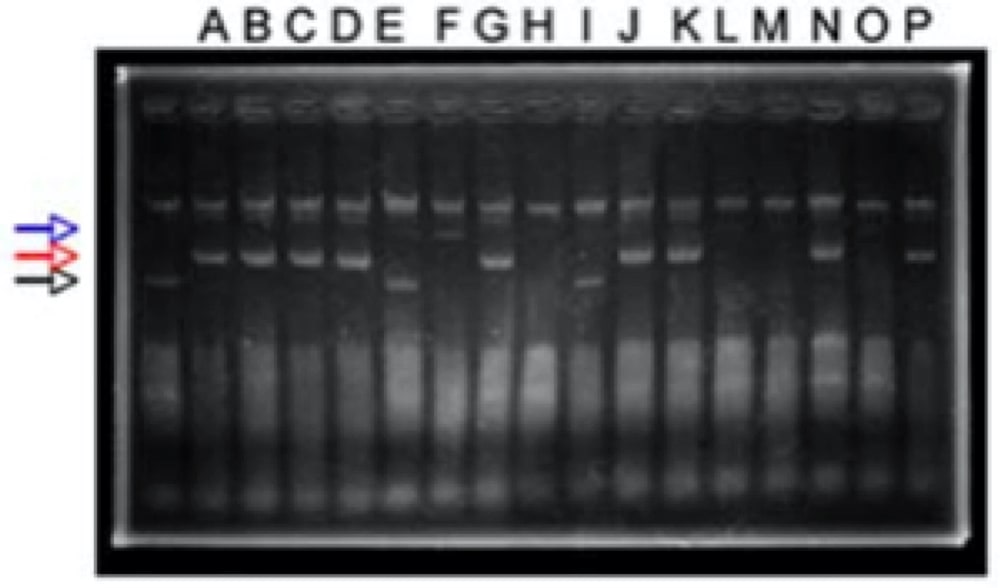
図3:左端のレーンがコントロールのサンプルで黒矢印のバンドが空ベクターのサイズを示す。目的の断片が挿入されたプラスミドは赤矢印の位置に(レーン A–D など)、セルフダイマーは青矢印の位置に(F など)泳動された。一番上のバンドはゲノム DNA、下には RNA が観察される。
