

概要
動物細胞で働く蛋白質は、一般に糖鎖修飾やプロセシングなどの複雑な翻訳後修飾が生理活性に必須である場合が多い。こうした複雑に細工が施された蛋白質をリコンビナントで大量に作るには、バクテリアの発現系よりも動物細胞を用いた発現系が向いている。最近では、大腸菌ほどではないにしてもパッケージ化が進み、誰でも簡便に利用できるようになっており、動物細胞を利用した蛋白質の立体構造解析は急速に発展しているところである。
本稿では動物細胞で発現させた糖蛋白質の解析事例として、筆者が大阪大学蛋白質研究所の高木淳一教授の研究室に在籍した際に行った、α5β1インテグリン細胞外ドメインと機能阻害抗体であるSG/19 Fab断片との複合体の精製、結晶化について詳述する。またそれに加えて、表題とは少し外れるが筆者らが苦労した抗体の一次配列決定法についても詳細を述べる。
イントロダクション
インテグリンは細胞接着に重要な役割を果たす分子であり、細胞外でコラーゲンやラミニン、フィブロネクチンなどの細胞外マトリックスと、細胞内でアクチンフィラメントなどの細胞骨格系とを繋いでいる(1)。その最も大きな特徴は、細胞外の情報を細胞内へ(outside-in signal)、細胞内の情報を細胞外へ(inside-out signal)と伝達する双方向性の受容体として働くことである。インテグリンはα鎖とβ鎖のヘテロ二量体から成り立っており、ヒトではα鎖が18種類、β鎖が8種類存在するがヘテロ二量体としての組み合わせは24種類に限定されている。この組み合わせの違いによって、インテグリンのリガンドである細胞外マトリックスに対する特異性などが異なっている。
インテグリンにはリガンドに対する親和性の低い状態(低親和性状態)と高い状態(高親和性状態)の二つの状態がある。これらの状態の切り替えメカニズムは”起き上がりモデル”として説明されている(2)。このモデルではリガンドに対する親和性の違いが細胞外ドメインの構造変化と相関しており、全体が大きく折れ曲がった状態が低親和性状態、全体が起き上がって足(α鎖とβ鎖のC末端側)が開いた状態が高親和性状態になっている。つまり、モジュラー構造をとっているインテグリンの細胞外ドメインは、細胞内外の刺激に応じてドメイン間の角度が変化し、リガンドに対する親和性が変化すると考えられているのである。
α5β1インテグリンは最も早く発見されたインテグリンで細胞外マトリックスであるフィブロネクチンの受容体である(3)。この二つの蛋白質の相互作用は、初期発生時の細胞移動の際に重要な役割を果たしており、α5β1インテグリン及びフィブロネクチンのノックアウトマウスは同じような表現型を示し、共に胚性致死である。このインテグリンは、抗β1抗体であるSG/19という抗体によってアロステリックにリガンド結合が阻害されることが知られている(4)。つまりドメイン間の角度を強制的に低親和性状態に固定させることによって、リガンド結合を妨げているのである。
今回、筆者らはα5β1インテグリン細胞外ドメインとSG/19 Fab断片との複合体のX線結晶構造解析に取り組み、2.9Å分解能での構造解析に成功した(5)。本稿ではその時に使用した実験プロトコールを余すところなく公開する。
装置・器具・試薬
装置
- 遠心機(ベックマンHP-30、ローターはJLA10.500)
- 精製装置(AKTAFPLC (GE Healthcare)、カラムはSuperdex 200 10/300 GL(GE Healthcare))
- 結晶化装置(微量分注装置としてMosquito(TTP Biotech)、プレートとしてMRC-2 plate(Hampton Research))
- その他SDSゲル電気泳動装置、クリーンベンチ、インキュベータ、ソニケーター、PCR装置など
器具
- ローラーボトル(Corning)
- エコノカラム(Bio-rad)
- 限外濾過濃縮器(Corning, Spin-X)
- 限外濾過濃縮器(少量)(Millipore, Bio-max)
- 透析チューブ(Spectra/Por Dialysis Tubing)
試薬
- Minimum Essential Medium (MEM) alpha (Invitrogen)
- Foetal bovine serum (EuroClone)
- MEM Non-essential amino acids (NEAA) 100x (Sigma)
- Sodium Pyruvate 100x (Gibco)
- Penicillin Streptmycin (Pen Strept) (Sigma)
- G418 sulfate (Calbiochem)
- Puromycin (Sigma)
- 硫酸アンモニウム(試薬特級)
- CNBr-activated sepharose 4B (GE Healthcare)
- rProteinA sepharose (GE Healthcare)
- Immobilized papain (Thermo Scientific)
- Antibody Binding and Elution Buffers (Thermo Scientific)
- L-システイン(試薬特級)
- SV40 Total RNA isolation (Promega)
- One-step RT-PCR kit (Qiagen)
- Ig-primer set (Novagen)
- PCR cloning kit (Qiagen)
実験手順
1)α5β1インテグリンの調製
2)SG/19 Fab断片の調製
3)α5β1インテグリン‐SG/19 Fab断片複合体の調製
4)α5β1インテグリン‐SG/19 Fab断片複合体の結晶化
5)SG/19 Fab断片の一次配列決定
実験の詳細
1)α5β1インテグリンの調製
本研究ではα5β1インテグリン細胞外ドメインのうちα5鎖についてはN末端から623番目までを、β1鎖についてはN末端から445番目までを使用している。形質転換体の作成に使う培養細胞は、CHO細胞のLec変異体であるCHO-lec3.2.8.1細胞を用いた。この細胞を用いて目的の糖蛋白質を発現させると、得られた糖蛋白質上のN型糖鎖もO型糖鎖も共に均一になることが知られている(6)。より詳細には、N型糖鎖の場合高マンノース型のMan5GlcNAc2という構造の糖鎖がアスパラギン残基の先に付加し、O型糖鎖の場合GalNAcの単糖のみがセリンもしくはスレオニンの先に付加される。α5β1インテグリンには多くの糖鎖修飾部位が存在し、今回デザインしたコンストラクトにもα5鎖に9箇所、β1鎖に8箇所のN型糖鎖修飾モチーフが存在する。これらの糖鎖修飾は、発現や活性に重要であることが既に調べられている7)。今回は、これらの二つの遺伝子を同時にCHO-lec3.2.8.1細胞に導入し、共発現する安定発現株を樹立している。安定発現株の樹立は限界希釈法を用いた。詳細な方法については蛋白質科学会アーカイブのプロトコール#050 8)と概ね同じである。培地はMEM alpha培地(MEM alpha + 5%(v/v) Foetal Bovine Serum + 1%(v/v) 100x sodium pyruvate + 1%(v/v) 100x MEM NEAA + 0.5%(v/v) Pen Strept)に選択マーカーとして0.5 mg/mL G418と5 μg/mL puromycinを添加したものを用いた。大量培養はローラーボトルを使用した。1本のローラーボトルにつき~107個の細胞を撒きこみ、300mLのMEM alpha培地を添加した。ローラーボトルは、37℃に設定したローラーインキュベータ中で毎分5回転程度の速度でゆっくりと転倒混和した。培地の交換は1週間に1度行い、1本のローラーボトルを2~3カ月間培養した。
今回用いているコンストラクトはインテグリン細胞外ドメイン全体からみるとN末端の半分に相当する。これらを何の工夫もなしに発現させると、容易に二つのサブユニットが乖離してしまう。そこでコンストラクト作成時の工夫として、α5鎖とβ1鎖の互いのC末端を結び付けて発現させることにした。具体的にはそれぞれのサブユニットのC末端側にそれぞれ人工的なロイシンジッパー様ヘリックスを付与し、α鎖側には酸性残基を多く配置させ(Acidic zipper; A-zip)、一方のβ鎖側には塩基性残基を多く配置させた(Basic zipper; B-zip)。さらに、これらの人工ヘリックスの一箇所に人工的なジスルフィド結合を導入することによって、サブユニット間を強固に固定させることでヘテロ二量体として安定に精製できるようにしている。こうしたバインダーを何も考えずに付加すると、肝心の目的蛋白質の構造そのものを崩してしまう危険がある。そのためアミノ酸配列をよく吟味したうえで何種類かのコンストラクトを作成し、発現量や蛋白質の安定性などを比較することで最適なデザインのコンストラクトを見つけなければならなかった。
精製はまず、安定発現株の培養上清2~3Lを終濃度50%で硫安沈澱し、遠心(~10,000g、20分間程度、4℃)によって上清と分離するところから始まる。得られた沈澱をbuffer A (20mM Tris-HCl (pH8.0), 150mM NaCl, 1mM CaCl2, 1mM MgCl2)で懸濁する。目安は1Lの培養上清に対して懸濁液が30mL程度になるようにしている。よく懸濁し溶け残りがないことを確認したら、3,000~3,500gで15分間遠心し、目に見えるゴミなどを除いておく。次の段階の精製は、α5β1インテグリンのうちβ1鎖のC末端に付与したB-zip配列に対する抗体(文献未発表)をCNBrセファロースに結合させた抗体レジンを使用している。懸濁液と抗体レジンをボトルに詰め、4℃の低温室内で毎分5回転程度の速度で2~3時間程度撹拌したのち、空のエコノカラムを用いて抗体レジンを回収する。レジンをbuffer Aによって5~10CV洗浄したのち、溶出はbuffer B(50mM tri-ethylamine(pH 11.4), 150mM NaCl, 1mM CaCl2)で行う(図1左側)。溶出画分は1.5mLを1画分として、合計5画分溶出する。溶出後は、2M Tris-HCl(pH8.0)溶液を1画分に50μL加えて速やかに中和してからbuffer Aに対して透析する。透析チューブは分子量カットオフで1万2千から4千程度のものを使用する。透析後は、限外濾過(分子量カットオフ10,000)によって1~2mg/mL程度になるように濃縮する。濃縮後はTEV proteaseを重量比で20分の1程度添加して、20℃、一昼夜かけてタグを切断する。切断の確認は、10%アクリルアミドゲルを用いたSDS電気泳動によって行う(図1右側)。筆者らは、この方法によって1Lの培養上清からα5β1インテグリン精製蛋白質を約1mg程度得ている。
インテグリンの発現、精製に関して幾つかのコメントを以下に述べる。
今回得られたα5β1インテグリンの糖鎖が、本当に均一であるかどうかの詳細な解析はしていない。しかし図1のSDS電気泳動のゲルを見る限り、それなりに均一であることがわかる。糖鎖修飾の経路に変異が入っていない細胞を使って糖蛋白質を発現させるともっとバンドが拡散する。
精製に関しては、タグの除去する際にTEV proteaseの切断効率がいつも100%にならず、やや切れ残ってしまった(図1右側及び図3なども参照)。実はTEV proteaseの代わりにエンドペプチダーゼであるAspNを用いると、TEV proteaseよりも高効率で、しかもより多くの余分な領域を除去することができる。筆者がこのことを見出した当初は、最終的な結晶の分解能の向上などを期待して大いに喜んだものだが、実際には分解能の向上には全く繋がらず逆に低下する結果となった。本研究で用いているSG/19はタグの有無に関わらずβ1に結合することができる。今回使用しているタグは各サブユニットそれぞれ40アミノ酸残基程度あり、さすがにこれを付けたままで結晶化を試したことはないのだが、ひょっとすると適度に切れ残ったタグが結晶の質に効いているのかもしれない。
また抗体カラムからインテグリンを溶出する溶液組成については、酸性条件で溶出するとインテグリン自体が変性し、しかも中性条件では溶出できなかったため、アルカリ条件で溶出することにした。
抗体からの目的蛋白質の溶出条件については、色々な溶液条件が知られおりそれらはThermo Scientificのホームページにまとめられている。興味のある方は参照して下さい。
(http://www.thermo.com/eThermo/CMA/PDFs/Articles/articlesFile_6645.pdf)
2)SG/19 Fab断片の調製
SG/19はマウス由来でサブクラスがIgG1に属している。SG/19 Fab断片の調製は、まずIgGの回収から始まる。SG/19ハイブリドーマの培養上清を回収し、遠心(3,000~3,500g, 15min, 4℃)によって細胞と上清とを分離したのち、NaClとTris-HCl (pH8.8)を終濃度でそれぞれ3.0Mと0.1Mになるように添加する。ポアサイズが0.44μmのフィルター濾過によってゴミなどを除いたのち、rProteinA sepharoseと混合し転倒混和する。筆者らは、ハイブリドーマの培養上清600mLに対してrProteinA sepharoseを、Bed volumeで10mL程度添加することを目安に使用している。混合溶液は空のエコノカラムを通すことでレジンだけを回収し、Thermo Scientific社製のBinding solution(成分非公開)で充分に洗浄し、Elution solution(成分非公開)によってIgGを溶出する。回収したIgGはPhosphate Buffered Saline (PBS)に対して透析し、限外濾過によって濃縮する。
ここからFab断片を精製するには、まず得られたIgGをPE buffer (20mM Na/K phosphate (pH7.0), 10mM EDTA)に対して透析し、濃縮したのちに、immobilized papain resinと混合する。そのままでは切断が進まないので終濃度250mMになるようにL-Cysteine溶液(pH7.0)を添加する。注意点としてはL-Cysteineをただ水に溶かすと酸性になるのでNaOHなどで中性になるように調製する。37℃に設定したインキュベータの中にローテータを入れ、resin混合溶液をゆっくりと撹拌する。筆者らは~30mgのIgGに対して、immobilized papain resinを600μL(Bed volume)程度使用し、3~5時間程度の反応時間で切断が完了するようにしている。切断の確認は15%アクリルアミドゲルを用いたSDS電気泳動で行う(図2左側)。反応溶液にはL-Cysteineが含まれているため、切断が適切に行われているかは還元条件下で比較する。切断反応が終了していることを確認した後、resin混合溶液を空のエコノカラムに通すことでpapain resinを分離し、素通り画分を集めてPBSに透析する(分子量カットオフ3,500)。透析をすると、L-Cysteineが沈澱して白く濁り、特有の臭いがすることがある。こうした際には、2~3時間おきに数回透析外液を交換しL-Cysteineが抜けきるように心掛ける。透析終了後は遠心(3,000~3,500g, 15min, 4℃)によって不溶物を除く。得られた上清は、等量のThermo Scientific社製のBinding solutionと混合し、エコノカラムに詰めたrProtein A sepharoseにかけ、素通り画分を回収する。このときのrProtein A sepharoseのBed volumeはIgGを精製した際と同量使用する。この素通り画分にFab断片が含まれているはずだが、念のためrProteinA sepharoseからElution solutionを用いて未反応のIgG及びFc断片を回収しておく。得られたFab断片はHBS buffer (20mM Hepes-NaOH (pH 7.0), 100mM NaCl)に対して透析し(分子量カットオフ3500)、濃縮したのちBCA法などの適当な方法を用いて蛋白質濃度を測定する。また精製純度は15%アクリルアミドゲルを用いたSDS電気泳動によって確認する(図2)。
SG/19 Fab断片の精製に関して幾つかのコメントを以下に述べる。
まず今回の精製にはrProtein A sepharoseを使用している。マウスのIgG1であれば、Protein G sepharoseを使用することも、勿論可能である。筆者らがrProtein Aを使用したのは、たまたま研究室にあったものを使用したところ上手く精製ができたからである。Bed volumeの目安も、培養上清600mLに対して10mL程度としたが、最適化の結果というよりはこのくらい大量にレジンを使用すれば漏れなく回収できると考えたからである。実際、筆者らは複数種類の抗体を同様な方法で精製した経験があるが、少なくともキャパシティーを超えてIgGなりFc断片が素通りしてしまったということはなかった。
抗体の精製に使用した溶液は、Thermo Scientific社から販売されている溶液セットをそのまま使用した。それらの大まかな成分は以下のURLに公開されている。今回、筆者らは精製に使用する溶液の最適化などは行わなかった。なぜなら常に不足するのはインテグリンの精製蛋白質の方であり、どんな方法をとっても抗体は相対的に大過剰でとれてしまうからである。従って、抗体の精製方法自体に関しては改良の余地は充分にあるかもしれない。
http://www.piercenet.com/browse.cfm?fldID=01010401
3)α5β1インテグリン‐SG/19 Fab断片複合体の調製
α5β1インテグリンとSG/19 Fab断片複合体の調製は、まず量比を決定するところから行う。精製したα5β1インテグリンとSG/19 Fab断片を少量ずつ分取し、量比を変えながら数回ゲル濾過カラム(Superdex200 10/300GL)にかけ、複合体の形成具合を見積もる。使用する溶液はbuffer C(20mM Tris-HCl (pH8.0), 100mM NaCl, 1mM CaCl2, 1mM MgCl2)である。筆者らの経験では、BCA法などから計算した蛋白質濃度を用いてSG/19 Fab断片の量とα5β1インテグリンの量を等量程度混合したのでは、SG/19 Fab断片の量が不足してしまった。SG/19 Fab断片は容易に大量調製が可能なため、思い切ってFab断片を5倍程度過剰に加えるようにしていた。(SG/19とβ1との結合親和性は報告されてはいないが、勿論これほど抗体過多にする必要はない。)得られたα5β1インテグリン-SG/19 Fab断片複合体は、SDS電気泳動で純度を確認し6~10mg/mL程度になるまで濃縮する(図3)。
SG/19 Fab断片は、精製直後にはSDS電気泳動の非還元条件で単一のバンドとして観察されたが、4℃で数日保存すると図3のように二つのバンドに分かれてしまった。幾種類かのプロテアーゼを用いて、より均一にしようと試みたものの状況は改善されなかった。いずれのサンプルを使っても結晶化には影響がなかったため、気にせずに用いることとした。
4)α5β1インテグリン‐SG/19 Fab断片複合体の結晶化
結晶化条件の初期スクリーニングは、スクリーニング試薬としてCrystal Screen, Index(Hampton Research), Precipitant Synergy(Emerald BioScience)を用いた。1回の精製サイクルで得られる蛋白質量は容量にして100μL程度と微量であったため、Mosquito (TTP biotech)という微量分注装置を用いて1条件辺り蛋白質と結晶化試薬をそれぞれ100nLずつ分注した。プレートはMRC-2の96ウェルプレートを使用し、シッティングドロップ蒸気拡散法にて行った。このときのリザーバー量は100μLとした。初期スクリーニング(全部で256条件)は精製ロット三回分を使用して行った。温度は20℃のみを検討した。また蛋白質濃度の検討は特に行わなかった。その結果、Indexにおける二つの条件で結晶が析出した。これらの条件は共にPEGを沈澱剤とし、中性のpH条件で、低塩濃度であった。インテグリンは一般にイオン強度を高くするとα鎖とβ鎖が乖離してしまうため、こうした条件が結晶の形成に適していたと考えられる。
結晶化条件の最適化は24-well plate(TPP)を用いてハンギングドロップ蒸気拡散法にて手作業で行った。ドロップは蛋白質溶液とリザーバー溶液をそれぞれ0.3μLずつカバーガラスの上で混合し、ウェルには500μLのリザーバー溶液を入れるようにした。研究当初、結晶の再現性が低く苦労したが、ミクロシーディング法により結晶核を導入することで再現性が飛躍的に向上した。
5)SG/19 Fab断片の一次配列決定
結晶構造解析には、当然のことながら結晶中に含まれている分子の一次配列情報を得ることが欠かせない。今回構造解析に使用したSG/19は一次配列が未知であったため、その一次配列を我々自身で決定する必要があった。筆者らはこのような作業に不慣れであったせいか、思いのほか苦労したので、ここに方法を記述する。
まずSG/19ハイブリドーマの培養液10mL分からSV40 Total RNA isolation (Promega社)を用いてクリーンベンチ内でmRNAを回収する。回収量は10mL分の細胞培養液から20~40μg程度のRNAが得られた。
次に逆転写PCR(RT-PCR)を行うのだが、使用するプライマーは、forward側はNovagenから販売されているIg-primer setに含まれるリーダー配列に対する縮重プライマーを使用した。またreverse側は抗体研究で著名なKabatのデータベースを参照し、IgGの重鎖(VH, CH1)と軽鎖(VL, CL)に関して網羅的に配列を調べて独自にデザインした。RT-PCRのサイクルとステップ数は以下の通り。
ステップ1;50℃、30分間\1サイクル
ステップ2;95℃、15分間\1サイクル
ステップ3;(94℃、1分間、50℃、1分間、72℃、1分間)\40サイクル
ステップ4;72℃、10分間\1サイクル
RT-PCRの結果、アガロース電気泳動により重鎖も軽鎖も順調に転写産物の増幅が見られたため、TA-cloningによってcDNAをpDrive vector (Qiagen)に組み込んだ。コロニーの青白判別によって目的遺伝子が組み込まれたベクターを選別し、塩基配列を決定した。得られた塩基配列と結晶中の電子密度を比較すると、重鎖は完全に一致したものの、軽鎖では全く電子密度と一致せず、しかも途中で終止コドンの入った偽の塩基配列しか得られなかった。RT-PCRの温度やサイクル数、使用する酵素などを検討したが改善が得られなかったため、SG/19蛋白質のN末端アミノ酸配列解析を行い、そこから縮重プライマーを設計し直すことにした。
まずSG/19 IgG蛋白質10μg程度を10%アクリルアミドゲルでSDS電気泳動に流し、それをPVDF膜に転写したのち、軽鎖のバンドを切り出してエドマン分解によるN末端アミノ酸配列解析を行った。得られた配列に対して図4のように三種類の縮重プライマーをデザインした。これらをforward側にして再度RT-PCRを行い、塩基配列決定をしたところ電子密度と完全に符合するアミノ酸配列を得ることができた。
筆者らはSG/19以外の抗体についても配列解析と構造解析の両方を行った経験があるのだが、電子密度に合わない偽配列が配列解析から得られることはままあるようである。我々のケースでは、得られた配列の妥当性が構造解析によって検証できるのだが、そうでない場合も充分あり得ることと思われる。こうした研究の難しさ、怖さを垣間見た思いである。
工夫とコツ
ミクロシーディング法のストック溶液の作成
今回の結晶構造解析は、ミクロシーディング法によって結晶化の再現性を向上させることが必須であった。そこで、コツとして筆者らが行ったミクロシーディング法の詳細を述べる。まず、結晶を数μLのリザーバーのドロップにナイロンループなどですくって移す。次に結晶をドロップ内で粉砕し、粉砕した破片をさらにリザーバー溶液を追加してピペッティングによってよく懸濁する。懸濁液を1.5mLのチューブに詰め、室温でソニケーター(Branson Model2510などの、いわゆる眼鏡洗浄機)で数十秒間程度ソニケーションを加えて、さらに細かく砕く。目安として、100x100x20μm程度の板状結晶を250μL程度のリザーバー溶液で懸濁する。この懸濁液をミクロシーディング溶液として、結晶化条件と同じ20℃に保存する。使用する際は、軽く指で叩くなどしてチューブ内を撹拌するようにした。時間的には結晶化ドロップを作成してから少なくとも1日程度経過していることを目安に、ドロップのカバーガラスを外しP2のピペットマンで結晶化ドロップの10分の1量程度を目安に添加した。ミクロシーディング溶液は20℃で少なくとも一カ月程度は保存可能だった。
縮重プライマーのデザイン
筆者らは一連の配列決定の作業について文献9などを参考にして行ったのであるが、そこでは縮重プライマーの組み合わせの総数は千以下になるようにするのが望ましいとあった。今回の場合でも、図4のプライマー①では128種類、②では768種類、③では3072種類の混合プライマーになるが、すべてで目的遺伝子の増幅が見られた。ただその際に3’末端側の配列(図4ではc)が一義的に決定されているのが重要であるようで、ここをn(4種類の混合)とすると全く遺伝子の増幅が見られなかった。
実験の安全
特に危険な作業などはありません。細胞の取り扱いはクリーンベンチ内で行いましょう。
謝辞
本研究は、筆者が大阪大学蛋白質研究所プロテオミクス総合研究センターに在籍した際に行ったものである。高木淳一教授には研究全般に渡って数多くのご指導、ご助言をいただいた。また結晶構造解析については禾晃和博士(現、横浜市立大学准教授)にお力添えをいただいた。その他、個々の実験に関しては、川上恵子さん、野田舞子さん、三原恵美子さん、清原丈嗣くんに助けていただいた。
文献
- Hynes Cell, 110, 673-87 (2002)
- Carman, C.V. & Springer, T.A., Curr.Opin.Cell Biol., 15, 547-56 (2003)
- Pytela, R. et al., Cell, 40, 191-8 (1985)
- Luo, B.H. et al., J.Biol.Chem. 279, 27466-71 (2004)
- Nagae et al., in preparation
- Stanley, P., Mol.Cell Biol., 9, 377-383 (1989)
- Isaji, T. et al., J.Biol.Chem. 281, 33258-33267 (2006)
- 浅野竜太郎, 蛋白質科学会アーカイブ, 2, e050 (2009)
- 中山広樹,バイオ実験イラストレイテッド,本当にふえるPCR,秀潤社
-
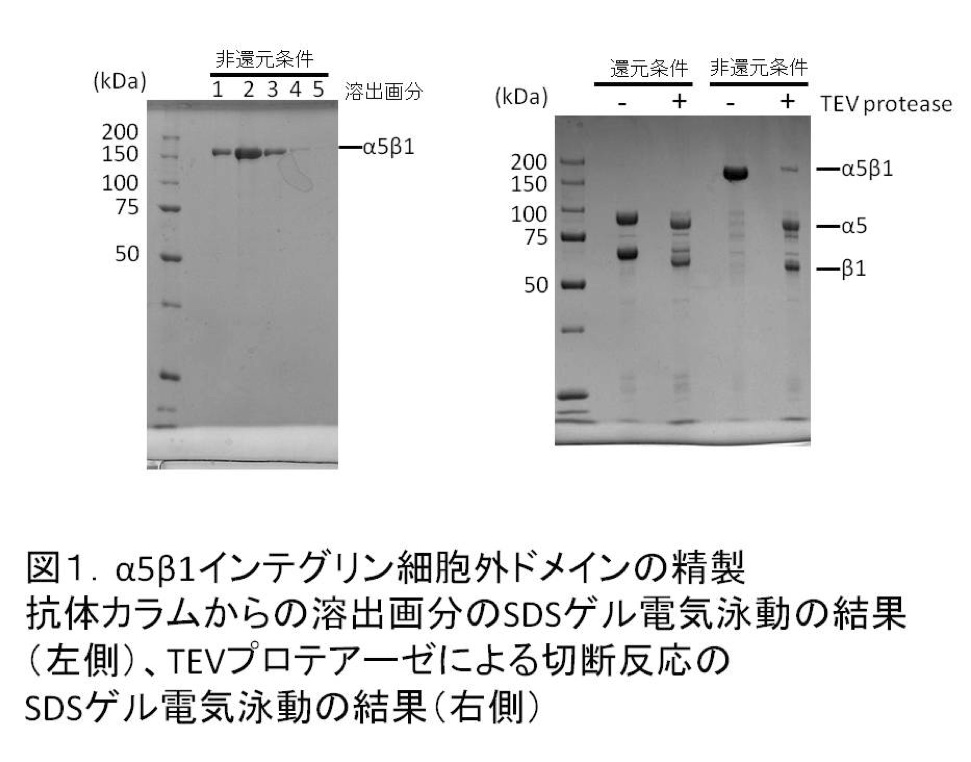
図1 -
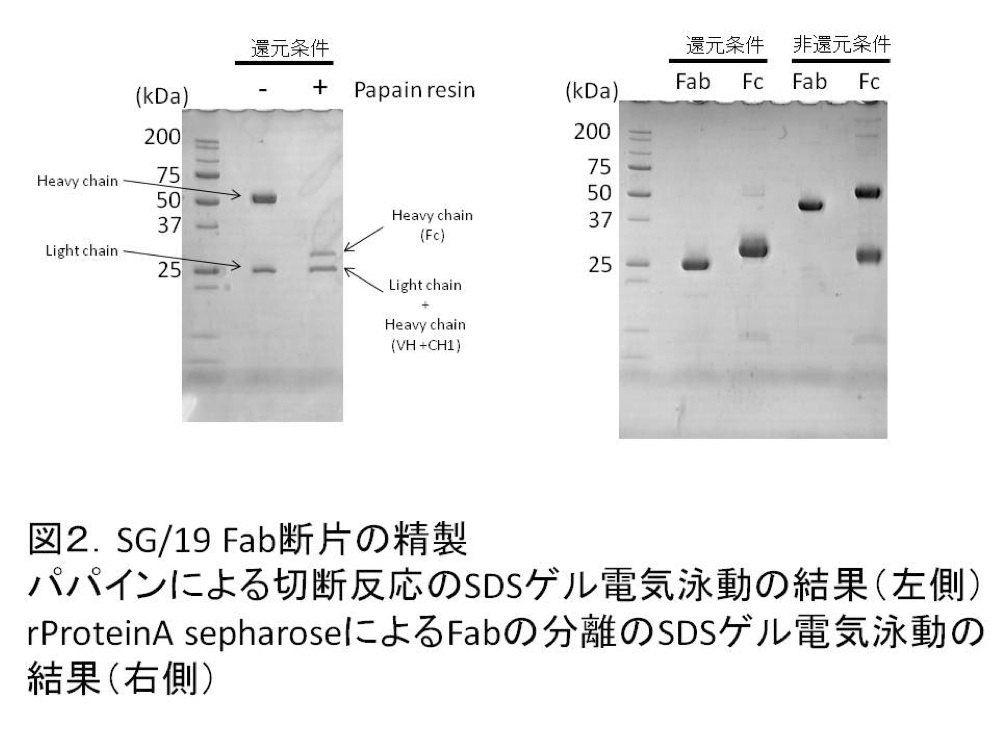
図2 -
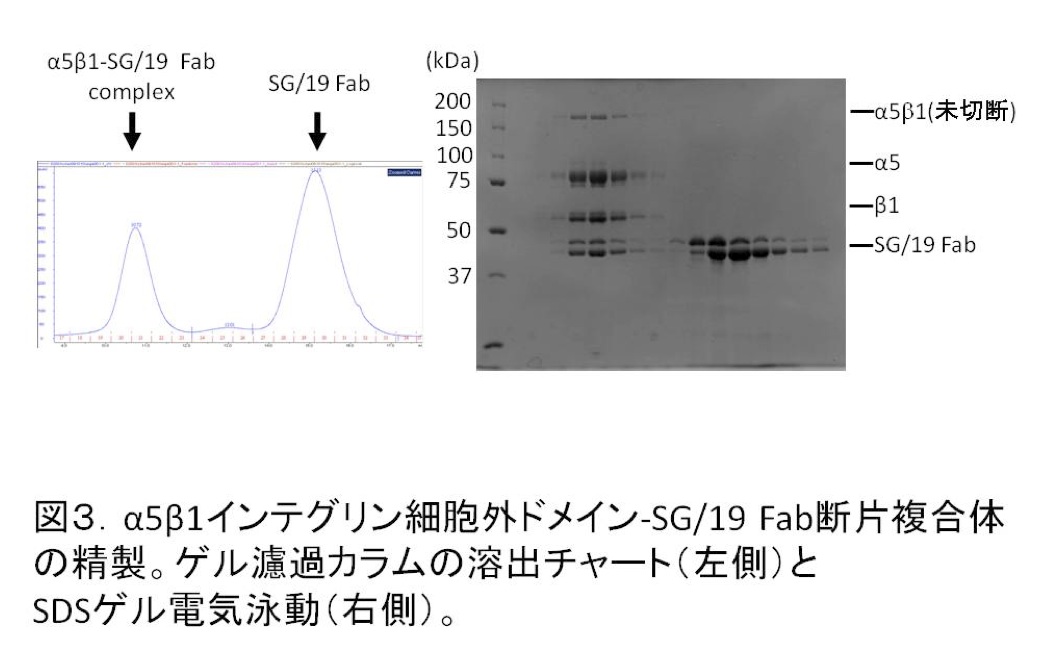
図3 -

図4
概要
動物細胞で働く蛋白質は、一般に糖鎖修飾やプロセシングなどの複雑な翻訳後修飾が生理活性に必須である場合が多い。こうした複雑に細工が施された蛋白質をリコンビナントで大量に作るには、バクテリアの発現系よりも動物細胞を用いた発現系が向いている。最近では、大腸菌ほどではないにしてもパッケージ化が進み、誰でも簡便に利用できるようになっており、動物細胞を利用した蛋白質の立体構造解析は急速に発展しているところである。
本稿では動物細胞で発現させた糖蛋白質の解析事例として、筆者が大阪大学蛋白質研究所の高木淳一教授の研究室に在籍した際に行った、α5β1インテグリン細胞外ドメインと機能阻害抗体であるSG/19 Fab断片との複合体の精製、結晶化について詳述する。またそれに加えて、表題とは少し外れるが筆者らが苦労した抗体の一次配列決定法についても詳細を述べる。
イントロダクション
インテグリンは細胞接着に重要な役割を果たす分子であり、細胞外でコラーゲンやラミニン、フィブロネクチンなどの細胞外マトリックスと、細胞内でアクチンフィラメントなどの細胞骨格系とを繋いでいる(1)。その最も大きな特徴は、細胞外の情報を細胞内へ(outside-in signal)、細胞内の情報を細胞外へ(inside-out signal)と伝達する双方向性の受容体として働くことである。インテグリンはα鎖とβ鎖のヘテロ二量体から成り立っており、ヒトではα鎖が18種類、β鎖が8種類存在するがヘテロ二量体としての組み合わせは24種類に限定されている。この組み合わせの違いによって、インテグリンのリガンドである細胞外マトリックスに対する特異性などが異なっている。
インテグリンにはリガンドに対する親和性の低い状態(低親和性状態)と高い状態(高親和性状態)の二つの状態がある。これらの状態の切り替えメカニズムは”起き上がりモデル”として説明されている(2)。このモデルではリガンドに対する親和性の違いが細胞外ドメインの構造変化と相関しており、全体が大きく折れ曲がった状態が低親和性状態、全体が起き上がって足(α鎖とβ鎖のC末端側)が開いた状態が高親和性状態になっている。つまり、モジュラー構造をとっているインテグリンの細胞外ドメインは、細胞内外の刺激に応じてドメイン間の角度が変化し、リガンドに対する親和性が変化すると考えられているのである。
α5β1インテグリンは最も早く発見されたインテグリンで細胞外マトリックスであるフィブロネクチンの受容体である(3)。この二つの蛋白質の相互作用は、初期発生時の細胞移動の際に重要な役割を果たしており、α5β1インテグリン及びフィブロネクチンのノックアウトマウスは同じような表現型を示し、共に胚性致死である。このインテグリンは、抗β1抗体であるSG/19という抗体によってアロステリックにリガンド結合が阻害されることが知られている(4)。つまりドメイン間の角度を強制的に低親和性状態に固定させることによって、リガンド結合を妨げているのである。
今回、筆者らはα5β1インテグリン細胞外ドメインとSG/19 Fab断片との複合体のX線結晶構造解析に取り組み、2.9Å分解能での構造解析に成功した(5)。本稿ではその時に使用した実験プロトコールを余すところなく公開する。
装置・器具・試薬
装置
- 遠心機(ベックマンHP-30、ローターはJLA10.500)
- 精製装置(AKTAFPLC (GE Healthcare)、カラムはSuperdex 200 10/300 GL(GE Healthcare))
- 結晶化装置(微量分注装置としてMosquito(TTP Biotech)、プレートとしてMRC-2 plate(Hampton Research))
- その他SDSゲル電気泳動装置、クリーンベンチ、インキュベータ、ソニケーター、PCR装置など
器具
- ローラーボトル(Corning)
- エコノカラム(Bio-rad)
- 限外濾過濃縮器(Corning, Spin-X)
- 限外濾過濃縮器(少量)(Millipore, Bio-max)
- 透析チューブ(Spectra/Por Dialysis Tubing)
試薬
- Minimum Essential Medium (MEM) alpha (Invitrogen)
- Foetal bovine serum (EuroClone)
- MEM Non-essential amino acids (NEAA) 100x (Sigma)
- Sodium Pyruvate 100x (Gibco)
- Penicillin Streptmycin (Pen Strept) (Sigma)
- G418 sulfate (Calbiochem)
- Puromycin (Sigma)
- 硫酸アンモニウム(試薬特級)
- CNBr-activated sepharose 4B (GE Healthcare)
- rProteinA sepharose (GE Healthcare)
- Immobilized papain (Thermo Scientific)
- Antibody Binding and Elution Buffers (Thermo Scientific)
- L-システイン(試薬特級)
- SV40 Total RNA isolation (Promega)
- One-step RT-PCR kit (Qiagen)
- Ig-primer set (Novagen)
- PCR cloning kit (Qiagen)
実験手順
1)α5β1インテグリンの調製
2)SG/19 Fab断片の調製
3)α5β1インテグリン‐SG/19 Fab断片複合体の調製
4)α5β1インテグリン‐SG/19 Fab断片複合体の結晶化
5)SG/19 Fab断片の一次配列決定
実験の詳細
1)α5β1インテグリンの調製
本研究ではα5β1インテグリン細胞外ドメインのうちα5鎖についてはN末端から623番目までを、β1鎖についてはN末端から445番目までを使用している。形質転換体の作成に使う培養細胞は、CHO細胞のLec変異体であるCHO-lec3.2.8.1細胞を用いた。この細胞を用いて目的の糖蛋白質を発現させると、得られた糖蛋白質上のN型糖鎖もO型糖鎖も共に均一になることが知られている(6)。より詳細には、N型糖鎖の場合高マンノース型のMan5GlcNAc2という構造の糖鎖がアスパラギン残基の先に付加し、O型糖鎖の場合GalNAcの単糖のみがセリンもしくはスレオニンの先に付加される。α5β1インテグリンには多くの糖鎖修飾部位が存在し、今回デザインしたコンストラクトにもα5鎖に9箇所、β1鎖に8箇所のN型糖鎖修飾モチーフが存在する。これらの糖鎖修飾は、発現や活性に重要であることが既に調べられている7)。今回は、これらの二つの遺伝子を同時にCHO-lec3.2.8.1細胞に導入し、共発現する安定発現株を樹立している。安定発現株の樹立は限界希釈法を用いた。詳細な方法については蛋白質科学会アーカイブのプロトコール#050 8)と概ね同じである。培地はMEM alpha培地(MEM alpha + 5%(v/v) Foetal Bovine Serum + 1%(v/v) 100x sodium pyruvate + 1%(v/v) 100x MEM NEAA + 0.5%(v/v) Pen Strept)に選択マーカーとして0.5 mg/mL G418と5 μg/mL puromycinを添加したものを用いた。大量培養はローラーボトルを使用した。1本のローラーボトルにつき~107個の細胞を撒きこみ、300mLのMEM alpha培地を添加した。ローラーボトルは、37℃に設定したローラーインキュベータ中で毎分5回転程度の速度でゆっくりと転倒混和した。培地の交換は1週間に1度行い、1本のローラーボトルを2~3カ月間培養した。
今回用いているコンストラクトはインテグリン細胞外ドメイン全体からみるとN末端の半分に相当する。これらを何の工夫もなしに発現させると、容易に二つのサブユニットが乖離してしまう。そこでコンストラクト作成時の工夫として、α5鎖とβ1鎖の互いのC末端を結び付けて発現させることにした。具体的にはそれぞれのサブユニットのC末端側にそれぞれ人工的なロイシンジッパー様ヘリックスを付与し、α鎖側には酸性残基を多く配置させ(Acidic zipper; A-zip)、一方のβ鎖側には塩基性残基を多く配置させた(Basic zipper; B-zip)。さらに、これらの人工ヘリックスの一箇所に人工的なジスルフィド結合を導入することによって、サブユニット間を強固に固定させることでヘテロ二量体として安定に精製できるようにしている。こうしたバインダーを何も考えずに付加すると、肝心の目的蛋白質の構造そのものを崩してしまう危険がある。そのためアミノ酸配列をよく吟味したうえで何種類かのコンストラクトを作成し、発現量や蛋白質の安定性などを比較することで最適なデザインのコンストラクトを見つけなければならなかった。
精製はまず、安定発現株の培養上清2~3Lを終濃度50%で硫安沈澱し、遠心(~10,000g、20分間程度、4℃)によって上清と分離するところから始まる。得られた沈澱をbuffer A (20mM Tris-HCl (pH8.0), 150mM NaCl, 1mM CaCl2, 1mM MgCl2)で懸濁する。目安は1Lの培養上清に対して懸濁液が30mL程度になるようにしている。よく懸濁し溶け残りがないことを確認したら、3,000~3,500gで15分間遠心し、目に見えるゴミなどを除いておく。次の段階の精製は、α5β1インテグリンのうちβ1鎖のC末端に付与したB-zip配列に対する抗体(文献未発表)をCNBrセファロースに結合させた抗体レジンを使用している。懸濁液と抗体レジンをボトルに詰め、4℃の低温室内で毎分5回転程度の速度で2~3時間程度撹拌したのち、空のエコノカラムを用いて抗体レジンを回収する。レジンをbuffer Aによって5~10CV洗浄したのち、溶出はbuffer B(50mM tri-ethylamine(pH 11.4), 150mM NaCl, 1mM CaCl2)で行う(図1左側)。溶出画分は1.5mLを1画分として、合計5画分溶出する。溶出後は、2M Tris-HCl(pH8.0)溶液を1画分に50μL加えて速やかに中和してからbuffer Aに対して透析する。透析チューブは分子量カットオフで1万2千から4千程度のものを使用する。透析後は、限外濾過(分子量カットオフ10,000)によって1~2mg/mL程度になるように濃縮する。濃縮後はTEV proteaseを重量比で20分の1程度添加して、20℃、一昼夜かけてタグを切断する。切断の確認は、10%アクリルアミドゲルを用いたSDS電気泳動によって行う(図1右側)。筆者らは、この方法によって1Lの培養上清からα5β1インテグリン精製蛋白質を約1mg程度得ている。
インテグリンの発現、精製に関して幾つかのコメントを以下に述べる。
今回得られたα5β1インテグリンの糖鎖が、本当に均一であるかどうかの詳細な解析はしていない。しかし図1のSDS電気泳動のゲルを見る限り、それなりに均一であることがわかる。糖鎖修飾の経路に変異が入っていない細胞を使って糖蛋白質を発現させるともっとバンドが拡散する。
精製に関しては、タグの除去する際にTEV proteaseの切断効率がいつも100%にならず、やや切れ残ってしまった(図1右側及び図3なども参照)。実はTEV proteaseの代わりにエンドペプチダーゼであるAspNを用いると、TEV proteaseよりも高効率で、しかもより多くの余分な領域を除去することができる。筆者がこのことを見出した当初は、最終的な結晶の分解能の向上などを期待して大いに喜んだものだが、実際には分解能の向上には全く繋がらず逆に低下する結果となった。本研究で用いているSG/19はタグの有無に関わらずβ1に結合することができる。今回使用しているタグは各サブユニットそれぞれ40アミノ酸残基程度あり、さすがにこれを付けたままで結晶化を試したことはないのだが、ひょっとすると適度に切れ残ったタグが結晶の質に効いているのかもしれない。
また抗体カラムからインテグリンを溶出する溶液組成については、酸性条件で溶出するとインテグリン自体が変性し、しかも中性条件では溶出できなかったため、アルカリ条件で溶出することにした。
抗体からの目的蛋白質の溶出条件については、色々な溶液条件が知られおりそれらはThermo Scientificのホームページにまとめられている。興味のある方は参照して下さい。
(http://www.thermo.com/eThermo/CMA/PDFs/Articles/articlesFile_6645.pdf)
2)SG/19 Fab断片の調製
SG/19はマウス由来でサブクラスがIgG1に属している。SG/19 Fab断片の調製は、まずIgGの回収から始まる。SG/19ハイブリドーマの培養上清を回収し、遠心(3,000~3,500g, 15min, 4℃)によって細胞と上清とを分離したのち、NaClとTris-HCl (pH8.8)を終濃度でそれぞれ3.0Mと0.1Mになるように添加する。ポアサイズが0.44μmのフィルター濾過によってゴミなどを除いたのち、rProteinA sepharoseと混合し転倒混和する。筆者らは、ハイブリドーマの培養上清600mLに対してrProteinA sepharoseを、Bed volumeで10mL程度添加することを目安に使用している。混合溶液は空のエコノカラムを通すことでレジンだけを回収し、Thermo Scientific社製のBinding solution(成分非公開)で充分に洗浄し、Elution solution(成分非公開)によってIgGを溶出する。回収したIgGはPhosphate Buffered Saline (PBS)に対して透析し、限外濾過によって濃縮する。
ここからFab断片を精製するには、まず得られたIgGをPE buffer (20mM Na/K phosphate (pH7.0), 10mM EDTA)に対して透析し、濃縮したのちに、immobilized papain resinと混合する。そのままでは切断が進まないので終濃度250mMになるようにL-Cysteine溶液(pH7.0)を添加する。注意点としてはL-Cysteineをただ水に溶かすと酸性になるのでNaOHなどで中性になるように調製する。37℃に設定したインキュベータの中にローテータを入れ、resin混合溶液をゆっくりと撹拌する。筆者らは~30mgのIgGに対して、immobilized papain resinを600μL(Bed volume)程度使用し、3~5時間程度の反応時間で切断が完了するようにしている。切断の確認は15%アクリルアミドゲルを用いたSDS電気泳動で行う(図2左側)。反応溶液にはL-Cysteineが含まれているため、切断が適切に行われているかは還元条件下で比較する。切断反応が終了していることを確認した後、resin混合溶液を空のエコノカラムに通すことでpapain resinを分離し、素通り画分を集めてPBSに透析する(分子量カットオフ3,500)。透析をすると、L-Cysteineが沈澱して白く濁り、特有の臭いがすることがある。こうした際には、2~3時間おきに数回透析外液を交換しL-Cysteineが抜けきるように心掛ける。透析終了後は遠心(3,000~3,500g, 15min, 4℃)によって不溶物を除く。得られた上清は、等量のThermo Scientific社製のBinding solutionと混合し、エコノカラムに詰めたrProtein A sepharoseにかけ、素通り画分を回収する。このときのrProtein A sepharoseのBed volumeはIgGを精製した際と同量使用する。この素通り画分にFab断片が含まれているはずだが、念のためrProteinA sepharoseからElution solutionを用いて未反応のIgG及びFc断片を回収しておく。得られたFab断片はHBS buffer (20mM Hepes-NaOH (pH 7.0), 100mM NaCl)に対して透析し(分子量カットオフ3500)、濃縮したのちBCA法などの適当な方法を用いて蛋白質濃度を測定する。また精製純度は15%アクリルアミドゲルを用いたSDS電気泳動によって確認する(図2)。
SG/19 Fab断片の精製に関して幾つかのコメントを以下に述べる。
まず今回の精製にはrProtein A sepharoseを使用している。マウスのIgG1であれば、Protein G sepharoseを使用することも、勿論可能である。筆者らがrProtein Aを使用したのは、たまたま研究室にあったものを使用したところ上手く精製ができたからである。Bed volumeの目安も、培養上清600mLに対して10mL程度としたが、最適化の結果というよりはこのくらい大量にレジンを使用すれば漏れなく回収できると考えたからである。実際、筆者らは複数種類の抗体を同様な方法で精製した経験があるが、少なくともキャパシティーを超えてIgGなりFc断片が素通りしてしまったということはなかった。
抗体の精製に使用した溶液は、Thermo Scientific社から販売されている溶液セットをそのまま使用した。それらの大まかな成分は以下のURLに公開されている。今回、筆者らは精製に使用する溶液の最適化などは行わなかった。なぜなら常に不足するのはインテグリンの精製蛋白質の方であり、どんな方法をとっても抗体は相対的に大過剰でとれてしまうからである。従って、抗体の精製方法自体に関しては改良の余地は充分にあるかもしれない。
http://www.piercenet.com/browse.cfm?fldID=01010401
3)α5β1インテグリン‐SG/19 Fab断片複合体の調製
α5β1インテグリンとSG/19 Fab断片複合体の調製は、まず量比を決定するところから行う。精製したα5β1インテグリンとSG/19 Fab断片を少量ずつ分取し、量比を変えながら数回ゲル濾過カラム(Superdex200 10/300GL)にかけ、複合体の形成具合を見積もる。使用する溶液はbuffer C(20mM Tris-HCl (pH8.0), 100mM NaCl, 1mM CaCl2, 1mM MgCl2)である。筆者らの経験では、BCA法などから計算した蛋白質濃度を用いてSG/19 Fab断片の量とα5β1インテグリンの量を等量程度混合したのでは、SG/19 Fab断片の量が不足してしまった。SG/19 Fab断片は容易に大量調製が可能なため、思い切ってFab断片を5倍程度過剰に加えるようにしていた。(SG/19とβ1との結合親和性は報告されてはいないが、勿論これほど抗体過多にする必要はない。)得られたα5β1インテグリン-SG/19 Fab断片複合体は、SDS電気泳動で純度を確認し6~10mg/mL程度になるまで濃縮する(図3)。
SG/19 Fab断片は、精製直後にはSDS電気泳動の非還元条件で単一のバンドとして観察されたが、4℃で数日保存すると図3のように二つのバンドに分かれてしまった。幾種類かのプロテアーゼを用いて、より均一にしようと試みたものの状況は改善されなかった。いずれのサンプルを使っても結晶化には影響がなかったため、気にせずに用いることとした。
4)α5β1インテグリン‐SG/19 Fab断片複合体の結晶化
結晶化条件の初期スクリーニングは、スクリーニング試薬としてCrystal Screen, Index(Hampton Research), Precipitant Synergy(Emerald BioScience)を用いた。1回の精製サイクルで得られる蛋白質量は容量にして100μL程度と微量であったため、Mosquito (TTP biotech)という微量分注装置を用いて1条件辺り蛋白質と結晶化試薬をそれぞれ100nLずつ分注した。プレートはMRC-2の96ウェルプレートを使用し、シッティングドロップ蒸気拡散法にて行った。このときのリザーバー量は100μLとした。初期スクリーニング(全部で256条件)は精製ロット三回分を使用して行った。温度は20℃のみを検討した。また蛋白質濃度の検討は特に行わなかった。その結果、Indexにおける二つの条件で結晶が析出した。これらの条件は共にPEGを沈澱剤とし、中性のpH条件で、低塩濃度であった。インテグリンは一般にイオン強度を高くするとα鎖とβ鎖が乖離してしまうため、こうした条件が結晶の形成に適していたと考えられる。
結晶化条件の最適化は24-well plate(TPP)を用いてハンギングドロップ蒸気拡散法にて手作業で行った。ドロップは蛋白質溶液とリザーバー溶液をそれぞれ0.3μLずつカバーガラスの上で混合し、ウェルには500μLのリザーバー溶液を入れるようにした。研究当初、結晶の再現性が低く苦労したが、ミクロシーディング法により結晶核を導入することで再現性が飛躍的に向上した。
5)SG/19 Fab断片の一次配列決定
結晶構造解析には、当然のことながら結晶中に含まれている分子の一次配列情報を得ることが欠かせない。今回構造解析に使用したSG/19は一次配列が未知であったため、その一次配列を我々自身で決定する必要があった。筆者らはこのような作業に不慣れであったせいか、思いのほか苦労したので、ここに方法を記述する。
まずSG/19ハイブリドーマの培養液10mL分からSV40 Total RNA isolation (Promega社)を用いてクリーンベンチ内でmRNAを回収する。回収量は10mL分の細胞培養液から20~40μg程度のRNAが得られた。
次に逆転写PCR(RT-PCR)を行うのだが、使用するプライマーは、forward側はNovagenから販売されているIg-primer setに含まれるリーダー配列に対する縮重プライマーを使用した。またreverse側は抗体研究で著名なKabatのデータベースを参照し、IgGの重鎖(VH, CH1)と軽鎖(VL, CL)に関して網羅的に配列を調べて独自にデザインした。RT-PCRのサイクルとステップ数は以下の通り。
ステップ1;50℃、30分間\1サイクル
ステップ2;95℃、15分間\1サイクル
ステップ3;(94℃、1分間、50℃、1分間、72℃、1分間)\40サイクル
ステップ4;72℃、10分間\1サイクル
RT-PCRの結果、アガロース電気泳動により重鎖も軽鎖も順調に転写産物の増幅が見られたため、TA-cloningによってcDNAをpDrive vector (Qiagen)に組み込んだ。コロニーの青白判別によって目的遺伝子が組み込まれたベクターを選別し、塩基配列を決定した。得られた塩基配列と結晶中の電子密度を比較すると、重鎖は完全に一致したものの、軽鎖では全く電子密度と一致せず、しかも途中で終止コドンの入った偽の塩基配列しか得られなかった。RT-PCRの温度やサイクル数、使用する酵素などを検討したが改善が得られなかったため、SG/19蛋白質のN末端アミノ酸配列解析を行い、そこから縮重プライマーを設計し直すことにした。
まずSG/19 IgG蛋白質10μg程度を10%アクリルアミドゲルでSDS電気泳動に流し、それをPVDF膜に転写したのち、軽鎖のバンドを切り出してエドマン分解によるN末端アミノ酸配列解析を行った。得られた配列に対して図4のように三種類の縮重プライマーをデザインした。これらをforward側にして再度RT-PCRを行い、塩基配列決定をしたところ電子密度と完全に符合するアミノ酸配列を得ることができた。
筆者らはSG/19以外の抗体についても配列解析と構造解析の両方を行った経験があるのだが、電子密度に合わない偽配列が配列解析から得られることはままあるようである。我々のケースでは、得られた配列の妥当性が構造解析によって検証できるのだが、そうでない場合も充分あり得ることと思われる。こうした研究の難しさ、怖さを垣間見た思いである。
工夫とコツ
ミクロシーディング法のストック溶液の作成
今回の結晶構造解析は、ミクロシーディング法によって結晶化の再現性を向上させることが必須であった。そこで、コツとして筆者らが行ったミクロシーディング法の詳細を述べる。まず、結晶を数μLのリザーバーのドロップにナイロンループなどですくって移す。次に結晶をドロップ内で粉砕し、粉砕した破片をさらにリザーバー溶液を追加してピペッティングによってよく懸濁する。懸濁液を1.5mLのチューブに詰め、室温でソニケーター(Branson Model2510などの、いわゆる眼鏡洗浄機)で数十秒間程度ソニケーションを加えて、さらに細かく砕く。目安として、100x100x20μm程度の板状結晶を250μL程度のリザーバー溶液で懸濁する。この懸濁液をミクロシーディング溶液として、結晶化条件と同じ20℃に保存する。使用する際は、軽く指で叩くなどしてチューブ内を撹拌するようにした。時間的には結晶化ドロップを作成してから少なくとも1日程度経過していることを目安に、ドロップのカバーガラスを外しP2のピペットマンで結晶化ドロップの10分の1量程度を目安に添加した。ミクロシーディング溶液は20℃で少なくとも一カ月程度は保存可能だった。
縮重プライマーのデザイン
筆者らは一連の配列決定の作業について文献9などを参考にして行ったのであるが、そこでは縮重プライマーの組み合わせの総数は千以下になるようにするのが望ましいとあった。今回の場合でも、図4のプライマー①では128種類、②では768種類、③では3072種類の混合プライマーになるが、すべてで目的遺伝子の増幅が見られた。ただその際に3’末端側の配列(図4ではc)が一義的に決定されているのが重要であるようで、ここをn(4種類の混合)とすると全く遺伝子の増幅が見られなかった。
実験の安全
特に危険な作業などはありません。細胞の取り扱いはクリーンベンチ内で行いましょう。
謝辞
本研究は、筆者が大阪大学蛋白質研究所プロテオミクス総合研究センターに在籍した際に行ったものである。高木淳一教授には研究全般に渡って数多くのご指導、ご助言をいただいた。また結晶構造解析については禾晃和博士(現、横浜市立大学准教授)にお力添えをいただいた。その他、個々の実験に関しては、川上恵子さん、野田舞子さん、三原恵美子さん、清原丈嗣くんに助けていただいた。
文献
- Hynes Cell, 110, 673-87 (2002)
- Carman, C.V. & Springer, T.A., Curr.Opin.Cell Biol., 15, 547-56 (2003)
- Pytela, R. et al., Cell, 40, 191-8 (1985)
- Luo, B.H. et al., J.Biol.Chem. 279, 27466-71 (2004)
- Nagae et al., in preparation
- Stanley, P., Mol.Cell Biol., 9, 377-383 (1989)
- Isaji, T. et al., J.Biol.Chem. 281, 33258-33267 (2006)
- 浅野竜太郎, 蛋白質科学会アーカイブ, 2, e050 (2009)
- 中山広樹,バイオ実験イラストレイテッド,本当にふえるPCR,秀潤社
