

概要
高等生物の核内には無数にRNAが存在し、その中から特定のRNAのみを選択し、核から細胞質に輸送する蛋白質Exporin-5の働きを構造学的に明らかにするために、Exportin-5/RanGTP/pre-microRNA複合体の結晶化を行った。Exportin-5を例に分子量100Kを超える蛋白質の大腸菌による大量発現系の構築と、蛋白質/RNA複合体の構造解析可能な結晶を得ることができた方法について紹介する。
装置、器具、試薬
ジャーファメンター(いわしや)、遠心機、ソニケーター、Ni-NTA agarose(Qiagen)、Glutathione Sepharose(GE Healthcare)、Superdex200(GE Healthcare)、BioLogic(BioRad)、Amicon Ultra 10K(Millipore)、NanoDrop(Thermo)
pre-microRNA((株)ジーンデザイン)(pre-microRNAは、二本鎖のステムとループ領域をもつRNAである。本研究で用いたRNAはヒトpre-microRNA30の二本鎖ステム部分のみである)。
実験手順
1)蛋白質の発現 Exportin-5、RanGTP
2)Exportin-5、RanGTPの粗精製
3)複合体形成と精製
4)結晶化
5)X線回折実験
詳細1 分子量100k以上の蛋白質の大腸菌システムを用いた大量発現方法
ヒト由来Exportin-5(136kDa)の大量発現系の構築
pQE60ベクター(C末端6xHis、アンピシリン耐性)(Qiagen)にExportin-5遺伝子を挿入し、大腸菌M15株(カナマイシン耐性)(Qiagen)に形質転換した。
当初、大腸菌の増殖が途中で止まったり、発現量が微量(0.2mg/10L培養)だったりした。大腸菌で発現させるには分子量が大きすぎるからかもしれない。形質転換した同じLBプレートのコロニーでも発現量に差がみられた。そこでコロニーを一つずつ2mL培養し、OD600=約0.7になった時に100μLを採取し-80℃で15%グリセロールストックした。残りの培養液は、OD600=1.0の時に0.1mMIPTGを加え20℃で12時間誘導処理し、2mLのサンプリングチューブで集菌した。ペレットを可溶化バッファ(50mM TrisHCl(pH7.4)、7mM β-mercaptoethanol、10mM imidazole、5% Glycerol、5mM MgCl2、500mM NaCl、0.1% Tween20)1mLで懸濁し、氷上で超音波処理後遠心分離した。1.5mLサンプリングチューブにNi-NTA agarose(40μL)をとり可溶化バッファで洗っておいた。可溶化した上清をNiレジンと30分間混合し遠心分離後、上澄み液を捨て可溶化バッファでNiレジンをよく洗った。遠心分離後、Niレジンを吸わないように注意しながら可溶化バッファはできるかぎり捨てた。Niレジンに電気泳動用の2×SDSサンプルバッファを添加し、電気泳動を行った。(図1)電気泳動の結果から大量発現するコロニーを選択しそのグリセロールストックから大量培養を行った。2mLのLB培地にグリセロールストック全量を加え37℃、2時間培養した。次にその培養液を200mLのLB培地に移し37℃4時間培養した。前培養液はすべて10Lジャーファメンターに移し37℃で培養した。(培地組成は下記の通り)大量培養中にOD600が0.6-0.7になったときに培養液を約5mL採取し700μLずつ分注し50%グリセロールを300μL加え15%グリセロースストックとした。OD600が0.95-1.0(ジャーファメンターでの培養をはじめてから5時間後くらいが目安。これ以上長く培養時間のかかるものは、発現量が低かった。)になると培地温度を20℃まで急激にさげ、誘導処理剤IPTGを0.1mM添加し12時間後に集菌した。
培地組成は、1% Polypepton 、0.5% Bacto Yeast Extract、0.5% NaClで前培養を行う。本培養は、10Lジャーファメンターを用い、前培養用の培地組成に10% Glycerolを添加する(培地は、10% Glycerolも一緒にオートクレーブ滅菌する。15min、121℃)。オートクレーブ滅菌後、温度が十分下がってからアンピシリンを終濃度が100μg/mL、カナマイシンを終濃度が、50μg/mLで加えた。
詳細2 蛋白質/RNA複合体形成と精製
Exportin-5、RanGTPは大腸菌でそれぞれを発現させ、粗精製を行っておく。粗精製を行う目的は、大腸菌由来のtRNAの除去である。なぜなら、Exportin-5はtRNAと結合可能で、Exportin-5/RanGTP/tRNAが一旦三者複合体を形成してしまうと、目的の複合体であるExportin-5/RanGTP/pre-microRNA複合体との分離が困難であるからである。
①Exportin-5の粗精製
Exportin-5を発現させた大腸菌wet重量30g(5L培養分)に可溶化バッファと0.1mM Pefabloc RC(Sigma Aldrich)、0.1% Tween20を加え全量で80mLにし、 氷冷しながら超音波破砕後遠心分離し、上清にNi-NTA agarose(Qiagen)を加え1時間4℃で撹拌した。可溶化バッファでオープンカラム(BioRad)にいれたNiレジンをよく洗い、可溶化バッファに80mM imidazoleを加えExportin-5を溶出する。溶出液(約35mL)を500mM NaCl含むバッファで平衡化したPhenyl Sepharoseとオープンカラムで混合し室温で30分置いた。100mM NaCl含むバッファ(pH7.4)で担体をよく洗い MilliQで溶出した。溶出容器に最終溶出液が50mM TrisHCl(pH7.4)、2mM MgCl2、2mM DTTになるようにあらかじめ添加しておいた(最終溶出量は、約30mL)(図2レーンa)。
蛋白質をカラムから溶出して回収するタイミングは、Bradford試薬の色の変化で判断した(500μL Bradford試薬に対して、10μLの蛋白溶液を加える)。
可溶化バッファ組成
50mM TrisHCl(pH7.4)
7mM β-mercaptoethanol
10mM imidazole
5% Glycerol
5mM MgCl2
500mM NaCl
Phenyl Sepeharose カラム用バッファ組成
50mM TrisHCl(pH7.4)
2mM DTT
5% Glycerol
2mM MgCl2
②RanGTPの粗精製
Ran(1-176)遺伝子(C末40アミノ酸欠損型)はGST切断サイト(TEV)を入れたpGEX6pベクターにいれた。宿主大腸菌は、BL21(DE3)pLysS(STRATEGENE)を用いた。
Ran(1-176)は、Ran遺伝子のC末を除去することによってGTP型になるという文献を参考にした(Exportin-5は、RanGTP結合下において基質と複合体を形成する)。
10L培養したRan(菌体wet重量60g)を可溶化バッファと0.1mM Pefabloc SC(Sigma Aldrich)で150mL以下に懸濁した。氷冷しながら超音波破砕後遠心処理し、上清と可溶化バッファで平衡化したGlutathione Sepharose(GE Healthcare)(ゲル5mL)を50mLコニカルチューブ2本に分けて入れ1時間4℃で撹拌した後にオープンカラムに移した後に、可溶化バッファで担体をよく洗浄した(NaClを含まないことがこの後行う複合体形成のためには重要である)(図2レーンb(注))。
(注)純度はよくないが、GST-RanGTPの粗精製の目的は、大腸菌由来tRNAの除去であり、複合体形成後の精製で純度はあがるためこのときの純度は気にしなかった。
可溶化バッファ
50mM TrisHCl(pH7.4)
2mM DTT
2mM MgCl2
5% Glycerol
③複合体形成
Exportin-5、RanGTPは、特にRanGTPは単体で可溶化すると不安定なために粗精製が終わったらすぐに複合体を形成させ安定化しなければならなかった。そこで各蛋白質の粗精製から複合体の形成までは、数時間以内に行った。
RanGTP(②)を吸着させたGlutathione Sepharoseのオープンカラムに粗精製の終わったExportin-5(①)とpre-microRNA(ジーンデザイン合成RNA)1mM GTP、 Recombinant RNasin Ribonuclease inhibitor(Promega)800unit(20μL)を素早く混ぜ、2時間半、4℃でおだやかに撹拌した。
2時間半後、カラムの中で、白い粘性のある糸くずのような凝集物が浮いているが、これは変性したExportin-5が主成分で、きれいなピペットの先でとり除いた。大腸菌で発現させたリコンビナントExportin-5は、約30%しか複合体活性をもたないことがわかった。
カラムをよく洗浄し、10mM 還元型グルタチオン(pH7.4)を含むバッファで溶出した。(図2レーンd)カラムを洗浄するバッファに還元型グルタチオンを10mMになるようにはかりとり、1M TrisHCl(pH8.8)でpH7.4になるように調整した。
溶出液(約50mL)は、Niアフィニティオープンカラムに添加し、1時間4℃で穏やかに撹拌した。カラムをよく洗浄した後は80mMimidazoleを含むバッファで溶出した。(図2レーンf)この溶出液(約20mL)を、濃縮膜(Amicon Ultra10K(Millipore))を用いて5mLまで濃縮した。濃縮したサンプルにTEVプロテアーゼ(Invitrogen)を20μL加え、4℃に一晩置いた(図2レーンg)。
次に、切断されたGSTおよび、凝集したものをExportin-5/RanGTP/pre-microRNA複合体から分離するためにSuperdexカラムを用いてゲル濾過HPLC(BioRad)を行った。Hiload 26/60 Superdex 200 pg(GE Healthcare)カラムを、ゲル濾過用バッファ(20mM TrisHCl(pH7.4)、2mM MgCl2、2mMDTT)で平衡化し、5mLの蛋白溶液を注入し、流速1mL/minでゲル濾過用バッファを流した。
280nmの吸光度(A280)と純度検定(SDS-PAGE)でExportin-5/RanGTP/pre-microRNA複合体のピークフラクションを回収し、濃縮膜Amicon Ultra 10k (Millipore)を用いて5-10mg/mLまで濃縮した(図2レーンh)。
詳細3 結晶化
高純度に精製されたExportin-5/RanGTP/pre-microRNA複合体溶液を用いて、ハンギングドロップ蒸気拡散法によって結晶化を行った。複合体の結晶化を行う場合、結晶中でも複合体を形成していること、できた結晶が構造解析可能な単結晶でなければならない。我々が初めて得た結晶は、pre-microRNAのみが解離したExportin-5/RanGTP二者複合体結晶で、構造解析不可能な多結晶であった(結晶の写真と回折実験の結果図3)。以下にpre-microRNAを含み、単結晶を得た方法について記す。
①Exportin-5/RanGTP 結晶からExportin-5/RanGTP/pre-miRNA結晶へ
二者複合体の結晶が得られたリザーバー溶液の条件は、8-10% PEG3350、100mM MgCl2、100mM DTT、5mM spermine 4HCl、100mM MES(pH6.5)だった。ドロップは、蛋白質溶液とリザーバー溶液を1:1で混合した。MgCl2の効果によってRNAが蛋白質から解離される。50mM MgCl2条件下でゲル濾過を行った結果、三者は解離することが明らかになった。
Mg濃度を 2mMまで下げ、解離しやすいpre-microRNAを結晶化サンプル溶液に、サンプル溶液中に含まれているpre-microRNAの1.2倍量になるようにpre-microRNAを加えた。Exportin-5/RanGTP/pre-microRNA三者複合体の結晶は得られたが、多結晶のため構造解析不可能であった。
三者複合体が得られたリザーバー溶液条件
7-8% PEG3350
2mMMgCl2
100mM DTT
2mM spermine 4HCl
100mM MES(pH6.5)
三者複合体が得られた蛋白質溶液条件
2mM MgCl2
2mM DTT
20mM TrisHCl(pH7.4)
0.01mM pre-microRNA(ジーンデザイン)
②多結晶から単結晶へ
(1)ストリークシーディング
得られた結晶を2~3個、結晶が溶けない溶液50μLに移しseedbeads(Hampton Research)にいれ、ボルテックスミキサーにかけ結晶を細かく砕いた。ストリークシーディングするためにタングステンのような細い針を、seedbeadsの溶液にいれ種結晶を付着させ、針先でドロップに一筋の線を書いた。シーディングを行うときのリザーバー溶液に含まれる沈殿剤(PEG3350)の濃度は、約1%下げた。
ストリークシーディングにより結晶のできる空間的な間隔は広くなった。しかし、多結晶であった。
(2)ハンギングドロップ蒸気拡散法の条件の最適化
ハンギングドロップ蒸気拡散法では、ドロップ(液相)、気相、リザーバー溶液(液相)の間で、各成分の濃度変化がおこり平衡状態になる。
平衡状態になるまで各成分の濃度変化により、各成分の化学ポテンシャルの変化がおこり、結晶核ができ結晶成長を促す。結晶成長中に様々な成分の化学ポテンシャルが変化するために、成長中の結晶に新しい別の結晶核ができ多結晶になっているのではないかと考えた。そこで、ドロップ、リザーバー溶液の沈殿剤を除く各成分の濃度を一定に保ち、沈殿剤の化学ポテンシャル変化によってのみ結晶化を行った。体積が一定でないため、濃度一定にすることは厳密には不可能である。しかし体積変化は微量なので、体積変化は無視することができる。各成分のどの濃度で結晶ができるのか、条件を最適化した(図4)。
蛋白質溶液 リザーバー溶液 2mM MgCl2 2mM MgCl2 2mM DTT 100mM DTT 20mM Tris HCl pH7.4 5mM spermine.4HCl 6-7% PEG3350 100mM MES-NaOH(pH6.5)
蛋白質溶液(最適化後) リザーバー溶液(最適化後)
2mM MgCl2 2mM MgCl2
50mM DTT 50mM DTT
2mM spermine.4HCl 2mM spermine.4HCl
20mM Tris HCl pH7.4 3-5% PEG3350
100mM MES-NaOH(pH6.5)
ドロップは、蛋白質溶液1μL、リザーバー溶液1μLを混合した。この溶液条件の最適化により、単結晶を得ることのできる確率が、約1%に向上した。
文献
- Okada,C. et al., Science 326,1275-8 (2009)
-

図1 -
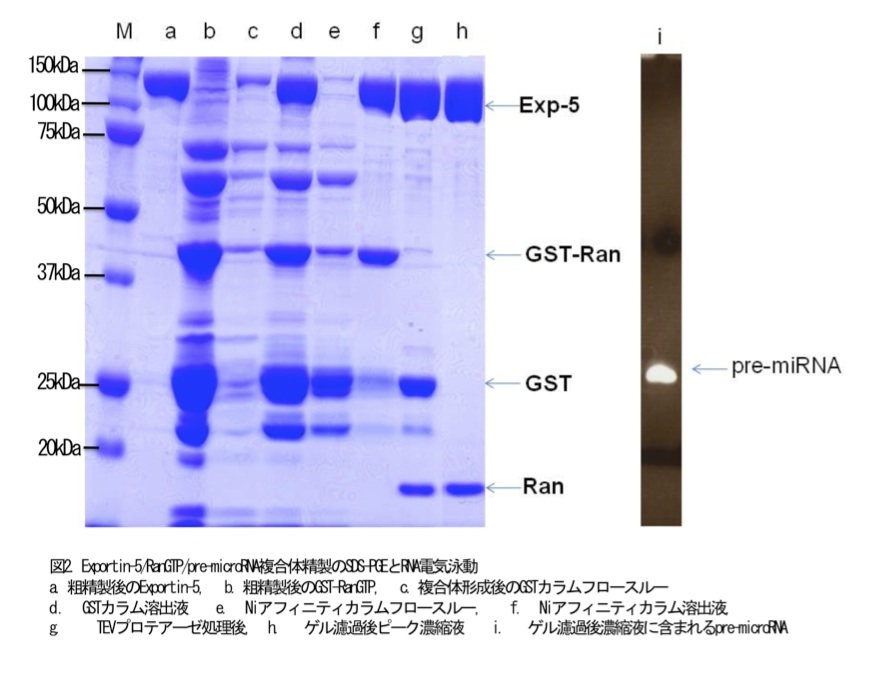
図2 -

図3 -

図4
概要
高等生物の核内には無数にRNAが存在し、その中から特定のRNAのみを選択し、核から細胞質に輸送する蛋白質Exporin-5の働きを構造学的に明らかにするために、Exportin-5/RanGTP/pre-microRNA複合体の結晶化を行った。Exportin-5を例に分子量100Kを超える蛋白質の大腸菌による大量発現系の構築と、蛋白質/RNA複合体の構造解析可能な結晶を得ることができた方法について紹介する。
装置、器具、試薬
ジャーファメンター(いわしや)、遠心機、ソニケーター、Ni-NTA agarose(Qiagen)、Glutathione Sepharose(GE Healthcare)、Superdex200(GE Healthcare)、BioLogic(BioRad)、Amicon Ultra 10K(Millipore)、NanoDrop(Thermo)
pre-microRNA((株)ジーンデザイン)(pre-microRNAは、二本鎖のステムとループ領域をもつRNAである。本研究で用いたRNAはヒトpre-microRNA30の二本鎖ステム部分のみである)。
実験手順
1)蛋白質の発現 Exportin-5、RanGTP
2)Exportin-5、RanGTPの粗精製
3)複合体形成と精製
4)結晶化
5)X線回折実験
詳細1 分子量100k以上の蛋白質の大腸菌システムを用いた大量発現方法
ヒト由来Exportin-5(136kDa)の大量発現系の構築
pQE60ベクター(C末端6xHis、アンピシリン耐性)(Qiagen)にExportin-5遺伝子を挿入し、大腸菌M15株(カナマイシン耐性)(Qiagen)に形質転換した。
当初、大腸菌の増殖が途中で止まったり、発現量が微量(0.2mg/10L培養)だったりした。大腸菌で発現させるには分子量が大きすぎるからかもしれない。形質転換した同じLBプレートのコロニーでも発現量に差がみられた。そこでコロニーを一つずつ2mL培養し、OD600=約0.7になった時に100μLを採取し-80℃で15%グリセロールストックした。残りの培養液は、OD600=1.0の時に0.1mMIPTGを加え20℃で12時間誘導処理し、2mLのサンプリングチューブで集菌した。ペレットを可溶化バッファ(50mM TrisHCl(pH7.4)、7mM β-mercaptoethanol、10mM imidazole、5% Glycerol、5mM MgCl2、500mM NaCl、0.1% Tween20)1mLで懸濁し、氷上で超音波処理後遠心分離した。1.5mLサンプリングチューブにNi-NTA agarose(40μL)をとり可溶化バッファで洗っておいた。可溶化した上清をNiレジンと30分間混合し遠心分離後、上澄み液を捨て可溶化バッファでNiレジンをよく洗った。遠心分離後、Niレジンを吸わないように注意しながら可溶化バッファはできるかぎり捨てた。Niレジンに電気泳動用の2×SDSサンプルバッファを添加し、電気泳動を行った。(図1)電気泳動の結果から大量発現するコロニーを選択しそのグリセロールストックから大量培養を行った。2mLのLB培地にグリセロールストック全量を加え37℃、2時間培養した。次にその培養液を200mLのLB培地に移し37℃4時間培養した。前培養液はすべて10Lジャーファメンターに移し37℃で培養した。(培地組成は下記の通り)大量培養中にOD600が0.6-0.7になったときに培養液を約5mL採取し700μLずつ分注し50%グリセロールを300μL加え15%グリセロースストックとした。OD600が0.95-1.0(ジャーファメンターでの培養をはじめてから5時間後くらいが目安。これ以上長く培養時間のかかるものは、発現量が低かった。)になると培地温度を20℃まで急激にさげ、誘導処理剤IPTGを0.1mM添加し12時間後に集菌した。
培地組成は、1% Polypepton 、0.5% Bacto Yeast Extract、0.5% NaClで前培養を行う。本培養は、10Lジャーファメンターを用い、前培養用の培地組成に10% Glycerolを添加する(培地は、10% Glycerolも一緒にオートクレーブ滅菌する。15min、121℃)。オートクレーブ滅菌後、温度が十分下がってからアンピシリンを終濃度が100μg/mL、カナマイシンを終濃度が、50μg/mLで加えた。
詳細2 蛋白質/RNA複合体形成と精製
Exportin-5、RanGTPは大腸菌でそれぞれを発現させ、粗精製を行っておく。粗精製を行う目的は、大腸菌由来のtRNAの除去である。なぜなら、Exportin-5はtRNAと結合可能で、Exportin-5/RanGTP/tRNAが一旦三者複合体を形成してしまうと、目的の複合体であるExportin-5/RanGTP/pre-microRNA複合体との分離が困難であるからである。
①Exportin-5の粗精製
Exportin-5を発現させた大腸菌wet重量30g(5L培養分)に可溶化バッファと0.1mM Pefabloc RC(Sigma Aldrich)、0.1% Tween20を加え全量で80mLにし、 氷冷しながら超音波破砕後遠心分離し、上清にNi-NTA agarose(Qiagen)を加え1時間4℃で撹拌した。可溶化バッファでオープンカラム(BioRad)にいれたNiレジンをよく洗い、可溶化バッファに80mM imidazoleを加えExportin-5を溶出する。溶出液(約35mL)を500mM NaCl含むバッファで平衡化したPhenyl Sepharoseとオープンカラムで混合し室温で30分置いた。100mM NaCl含むバッファ(pH7.4)で担体をよく洗い MilliQで溶出した。溶出容器に最終溶出液が50mM TrisHCl(pH7.4)、2mM MgCl2、2mM DTTになるようにあらかじめ添加しておいた(最終溶出量は、約30mL)(図2レーンa)。
蛋白質をカラムから溶出して回収するタイミングは、Bradford試薬の色の変化で判断した(500μL Bradford試薬に対して、10μLの蛋白溶液を加える)。
可溶化バッファ組成
50mM TrisHCl(pH7.4)
7mM β-mercaptoethanol
10mM imidazole
5% Glycerol
5mM MgCl2
500mM NaCl
Phenyl Sepeharose カラム用バッファ組成
50mM TrisHCl(pH7.4)
2mM DTT
5% Glycerol
2mM MgCl2
②RanGTPの粗精製
Ran(1-176)遺伝子(C末40アミノ酸欠損型)はGST切断サイト(TEV)を入れたpGEX6pベクターにいれた。宿主大腸菌は、BL21(DE3)pLysS(STRATEGENE)を用いた。
Ran(1-176)は、Ran遺伝子のC末を除去することによってGTP型になるという文献を参考にした(Exportin-5は、RanGTP結合下において基質と複合体を形成する)。
10L培養したRan(菌体wet重量60g)を可溶化バッファと0.1mM Pefabloc SC(Sigma Aldrich)で150mL以下に懸濁した。氷冷しながら超音波破砕後遠心処理し、上清と可溶化バッファで平衡化したGlutathione Sepharose(GE Healthcare)(ゲル5mL)を50mLコニカルチューブ2本に分けて入れ1時間4℃で撹拌した後にオープンカラムに移した後に、可溶化バッファで担体をよく洗浄した(NaClを含まないことがこの後行う複合体形成のためには重要である)(図2レーンb(注))。
(注)純度はよくないが、GST-RanGTPの粗精製の目的は、大腸菌由来tRNAの除去であり、複合体形成後の精製で純度はあがるためこのときの純度は気にしなかった。
可溶化バッファ
50mM TrisHCl(pH7.4)
2mM DTT
2mM MgCl2
5% Glycerol
③複合体形成
Exportin-5、RanGTPは、特にRanGTPは単体で可溶化すると不安定なために粗精製が終わったらすぐに複合体を形成させ安定化しなければならなかった。そこで各蛋白質の粗精製から複合体の形成までは、数時間以内に行った。
RanGTP(②)を吸着させたGlutathione Sepharoseのオープンカラムに粗精製の終わったExportin-5(①)とpre-microRNA(ジーンデザイン合成RNA)1mM GTP、 Recombinant RNasin Ribonuclease inhibitor(Promega)800unit(20μL)を素早く混ぜ、2時間半、4℃でおだやかに撹拌した。
2時間半後、カラムの中で、白い粘性のある糸くずのような凝集物が浮いているが、これは変性したExportin-5が主成分で、きれいなピペットの先でとり除いた。大腸菌で発現させたリコンビナントExportin-5は、約30%しか複合体活性をもたないことがわかった。
カラムをよく洗浄し、10mM 還元型グルタチオン(pH7.4)を含むバッファで溶出した。(図2レーンd)カラムを洗浄するバッファに還元型グルタチオンを10mMになるようにはかりとり、1M TrisHCl(pH8.8)でpH7.4になるように調整した。
溶出液(約50mL)は、Niアフィニティオープンカラムに添加し、1時間4℃で穏やかに撹拌した。カラムをよく洗浄した後は80mMimidazoleを含むバッファで溶出した。(図2レーンf)この溶出液(約20mL)を、濃縮膜(Amicon Ultra10K(Millipore))を用いて5mLまで濃縮した。濃縮したサンプルにTEVプロテアーゼ(Invitrogen)を20μL加え、4℃に一晩置いた(図2レーンg)。
次に、切断されたGSTおよび、凝集したものをExportin-5/RanGTP/pre-microRNA複合体から分離するためにSuperdexカラムを用いてゲル濾過HPLC(BioRad)を行った。Hiload 26/60 Superdex 200 pg(GE Healthcare)カラムを、ゲル濾過用バッファ(20mM TrisHCl(pH7.4)、2mM MgCl2、2mMDTT)で平衡化し、5mLの蛋白溶液を注入し、流速1mL/minでゲル濾過用バッファを流した。
280nmの吸光度(A280)と純度検定(SDS-PAGE)でExportin-5/RanGTP/pre-microRNA複合体のピークフラクションを回収し、濃縮膜Amicon Ultra 10k (Millipore)を用いて5-10mg/mLまで濃縮した(図2レーンh)。
詳細3 結晶化
高純度に精製されたExportin-5/RanGTP/pre-microRNA複合体溶液を用いて、ハンギングドロップ蒸気拡散法によって結晶化を行った。複合体の結晶化を行う場合、結晶中でも複合体を形成していること、できた結晶が構造解析可能な単結晶でなければならない。我々が初めて得た結晶は、pre-microRNAのみが解離したExportin-5/RanGTP二者複合体結晶で、構造解析不可能な多結晶であった(結晶の写真と回折実験の結果図3)。以下にpre-microRNAを含み、単結晶を得た方法について記す。
①Exportin-5/RanGTP 結晶からExportin-5/RanGTP/pre-miRNA結晶へ
二者複合体の結晶が得られたリザーバー溶液の条件は、8-10% PEG3350、100mM MgCl2、100mM DTT、5mM spermine 4HCl、100mM MES(pH6.5)だった。ドロップは、蛋白質溶液とリザーバー溶液を1:1で混合した。MgCl2の効果によってRNAが蛋白質から解離される。50mM MgCl2条件下でゲル濾過を行った結果、三者は解離することが明らかになった。
Mg濃度を 2mMまで下げ、解離しやすいpre-microRNAを結晶化サンプル溶液に、サンプル溶液中に含まれているpre-microRNAの1.2倍量になるようにpre-microRNAを加えた。Exportin-5/RanGTP/pre-microRNA三者複合体の結晶は得られたが、多結晶のため構造解析不可能であった。
三者複合体が得られたリザーバー溶液条件
7-8% PEG3350
2mMMgCl2
100mM DTT
2mM spermine 4HCl
100mM MES(pH6.5)
三者複合体が得られた蛋白質溶液条件
2mM MgCl2
2mM DTT
20mM TrisHCl(pH7.4)
0.01mM pre-microRNA(ジーンデザイン)
②多結晶から単結晶へ
(1)ストリークシーディング
得られた結晶を2~3個、結晶が溶けない溶液50μLに移しseedbeads(Hampton Research)にいれ、ボルテックスミキサーにかけ結晶を細かく砕いた。ストリークシーディングするためにタングステンのような細い針を、seedbeadsの溶液にいれ種結晶を付着させ、針先でドロップに一筋の線を書いた。シーディングを行うときのリザーバー溶液に含まれる沈殿剤(PEG3350)の濃度は、約1%下げた。
ストリークシーディングにより結晶のできる空間的な間隔は広くなった。しかし、多結晶であった。
(2)ハンギングドロップ蒸気拡散法の条件の最適化
ハンギングドロップ蒸気拡散法では、ドロップ(液相)、気相、リザーバー溶液(液相)の間で、各成分の濃度変化がおこり平衡状態になる。
平衡状態になるまで各成分の濃度変化により、各成分の化学ポテンシャルの変化がおこり、結晶核ができ結晶成長を促す。結晶成長中に様々な成分の化学ポテンシャルが変化するために、成長中の結晶に新しい別の結晶核ができ多結晶になっているのではないかと考えた。そこで、ドロップ、リザーバー溶液の沈殿剤を除く各成分の濃度を一定に保ち、沈殿剤の化学ポテンシャル変化によってのみ結晶化を行った。体積が一定でないため、濃度一定にすることは厳密には不可能である。しかし体積変化は微量なので、体積変化は無視することができる。各成分のどの濃度で結晶ができるのか、条件を最適化した(図4)。
蛋白質溶液 リザーバー溶液 2mM MgCl2 2mM MgCl2 2mM DTT 100mM DTT 20mM Tris HCl pH7.4 5mM spermine.4HCl 6-7% PEG3350 100mM MES-NaOH(pH6.5)
蛋白質溶液(最適化後) リザーバー溶液(最適化後)
2mM MgCl2 2mM MgCl2
50mM DTT 50mM DTT
2mM spermine.4HCl 2mM spermine.4HCl
20mM Tris HCl pH7.4 3-5% PEG3350
100mM MES-NaOH(pH6.5)
ドロップは、蛋白質溶液1μL、リザーバー溶液1μLを混合した。この溶液条件の最適化により、単結晶を得ることのできる確率が、約1%に向上した。
文献
- Okada,C. et al., Science 326,1275-8 (2009)
