

概要
放射光を利用したX線結晶構造解析は、その迅速性、精度の高さ、微小試料への適用性などの特徴から、今や構造生物学研究に欠かせないものとなっている。最新のビームラインでは、標準的な結晶であれば10分以内に1データセットを収集することができ、多波長異常分散法(MAD法)でも30分以内で測定完了することが可能であり、迅速X線回折実験が現実のものとなっている。また、データ精度の向上により、以前は困難であったイオウやリンなどの軽元素を利用した単波長異常分散法(SAD法)も適用可能となってきている。最近では、高輝度アンジュレータ光を光源とした微小集光ビームを広範に利用することができるようになっており、結晶性さえ良ければ10ミクロンを切るような試料でも構造解析可能となってきている。
これら回折実験の操作は、直感的なグラフィカルインターフェースで全て行なうことができ、誰でもすぐに実験可能なように整備されている。ロボット技術や計算機技術を利用した、測定の自動化に向けた開発も進められえており、近い将来には試料だけ放射光施設に送り、実験操作はそれぞれの研究室から直接行なったり、自動的にデータ測定が進められたりできるようになる予定である。
本プロトコールでは、放射光を利用した蛋白質結晶のX線回折実験について標準的な手法を概説し、今後どのような開発が進められるかについて、フォトンファクトリー(PF)の構造生物ビームラインを例として述べる。
装置・器具・試薬
回折実験は放射光施設に付属のビームライン及び実験装置で行なうため、特にユーザーが持ち込む装置はない(図1)。結晶化プレートや、予め凍結した試料をビームラインに持ち込み、実験装置にマウントすれば回折実験を開始することができる。凍結試料持込にはドライシッパー(内部に液体窒素吸着剤を充填し、倒れても液体窒素が流出しない特別な運搬用保存容器:例えばアメリカ「テイラーウォートン」社CXRシリーズ)を利用する。
マウント方法についても、大きな形状変化が無い限り、クライオループでもキャピラリーでも実験可能である。ただし、付属のサンプル交換ロボットを使用する際には、マウント方法が制限されるので注意する必要がある。例えば、PFの場合には、Hampton Research社(1)のCrystalCap Copper Magnetic 18mmにMounted Cryoloopのループに一番近い線で折ったループを挿したものを標準ピンとしており、その形状から大きく外れる場合には使用することができない(2)。また、カセットもCrystal Positioning Systems社のものを標準としており、その他のタイプのカセットは使用できない(UniPuckに関しては近い将来対応予定)。
実験手順
1)放射光施設への結晶試料運搬
2)回折計への結晶試料マウント
3)スナップショット(評価用回折像)測定
4)回折強度データ収集
4’)重原子を用いた異常分散実験(MAD法とSAD法)
4”)軽原子を用いた低エネルギーSAD実験
4”’)超高分解能回折データ測定
5)データ処理及び保存
実験の詳細
1)放射光施設への結晶試料運搬
蛋白質結晶は溶媒含有量が多く、周辺環境の変化に敏感なため、結晶試料の運搬は最も気を使う作業の一つである。試料は結晶化プレートのまま、もしくは母液バッチやガラスキャピラリー中に保管し、発泡スチロール容器などで温度変化が生じないようにして静かに運搬する。最近では、凍結させてから運搬することが一般的となってきており、ドライシッパーごと宅配便で直接放射光施設に送付することができる。その場合には、結晶試料をナイロン等のクライオループで抗凍結剤入りの母液ごと掬い凍結させ、ループごとケーンに固定して運搬する。試料交換ロボットを利用する際には、ループは専用のカセットに装填されなければならない。現在は各放射光施設で異なるカセットを利用しているが、共通に利用できるような開発が進められている。
2)回折計への結晶試料マウント
運搬された結晶試料を回折計の回転軸にマウントし、ビーム位置(回転中心)に正確に位置決めする。近年放射光施設では高輝度化が進み、通常は凍結試料でなければ放射線損傷のためデータ収集をすることができない。そのため、多くの場合回転軸には磁石ベースが準備されており、クライオループを磁石で固定してマウントする。ただしキャピラリーの場合でも、金属ベースを取り付けることにより簡便にマウントすることができる。
結晶試料をその場で凍結させる場合には、まず結晶化プレートや母液バッチからクライオループで掬う。母液には、氷形成による結晶損傷を防ぐため、多くの場合抗凍結剤(例えばグリセロールやスクロース等)を溶かしておく必要がある。そして、吹き付け低温装置の気流(窒素もしくはヘリウム)を一旦遮り、磁石ベースにマウントした後、なるべく短時間で吹き付けを再開する。気流を一旦遮るためのシャッターを準備している所も多い。凍結済みの試料をマウントする際には、専用の道具(クライオトング、ワンド、クランプ等)を使用して保存容器中のケーンから液体窒素中で試料を取り出し、大気に触れないように素早くマウントする。試料交換ロボットを使用する場合には、保存容器中の専用カセットをロボットの液体窒素デュワーにセットすれば、後はGUI操作でマウントすることができる。また、その場合には、カセットのどの位置にどういうサンプルが入っているかを、予めファイル(PFの場合にはCSVファイル、図2に記録しておくことで容易に試料管理をすることができる。後述する遠隔実験では必須の機能である。
マウントされた試料の位置決めは、GUI上で結晶を確認し、その位置を選択することにより行なう(図3)。90度回転させて同じ操作をすることにより、回転中心(ビーム位置)に正確に位置決めすることができる。最近では、画像解析技術を使ってループを認識して自動で位置決めする機能も装備されている。また、ビームスキャンをすることにより試料位置を判別する機能も開発されつつあり、近い将来には試料交換ロボットと併せて、全自動で試料交換が可能なシステムが実現するであろう。
3)スナップショット(評価用回折像)測定
結晶試料の位置決めが完了したら、結晶性や測定条件の評価のためのスナップショット撮影を行う(複合体サーチ等性質が分かっている結晶の場合には省略する場合もある)。測定した回折像は画像表示ソフトを利用して、分解能、回折点の分離、モザイク性などの評価を行うとともに、回折強度の統計を確認して適切な露光時間の見積もり、結晶方位や格子定数の確認から振動範囲や振動角の見積もりなど、測定条件の決定を行なう。PFの場合には、結晶性の評価にはadxvを利用しており、最大強度とオーバーロードの数を見ながら露光時間を調整している(図4)。一般的には、最大強度が飽和強度ギリギリ、もしくは多少のオーバーロード反射(数十ピクセル以内)が観測される程度に露光時間を設定すると良いデータが得られる。しかし一方で、露光時間を増加させると放射線損傷の問題が生じてくる。放射線損傷を受け易い結晶の場合には、測定できる分解能を下げてでも完全データセットを測定した方が良いであろう。最近では、RADDOSE(3)等の解析ソフトを用い、最大許容照射線量を見積もり、必要枚数で割ることで一枚当たりの露光時間を自動的に設定してくれる機能を装備しているインターフェースもある。また、振動範囲や振動角の見積もりには、データ処理ソフト(HKL2000(4)やmosflm(5)など)のstrategy機能を利用することが多い。
4)回折強度データ収集
結晶性評価をクリアして測定条件が決まれば、いよいよ回折強度データ収集となる。必要な領域を振動写真法で測定する。測定時間はビームラインによって異なるが、最近の挿入光源ビームラインでは、典型的な条件(1度振動、1秒露光、180枚測定)だと10分以内で測定が完了する。類似構造を利用できる場合には、良いデータセットを一つ測定できれば、これだけで構造解析をすることができる。
新規構造の場合には、複数のデータセットを参照して、新規の位相情報を求める必要がある。多重重原子同型置換法の場合、複数の重原子置換体結晶のデータセットを測定し、それらを組み合わせて構造解析を行なう。十分な位相情報が得られる同型の重原子置換体結晶を調製するためには、慎重な調製条件との検討と多くの結晶性評価実験を行わなければならなく多大な労力と時間がかかる。そこで最近では、放射光の波長可変性を積極的に利用し、異常分散効果により位相情報を引き出す方法が用いられることが多くなっている。この実験方法について以下に別項として述べる。
4’)重原子を用いた異常分散実験(MAD法とSAD法)
蛋白質の新規構造決定には、多重重原子同型置換法を利用することが多かったが、放射光の汎用利用と、遺伝子工学的にセレンを導入できるようになってから、重原子の異常分散効果を利用した位相決定法が広く用いられるようになった。
多波長異常分散法(MAD法)は、一つの結晶から吸収端近傍での異なる波長(通常3波長)でデータ収集を行い、それらデータセット間の異常分散差を利用して位相決定する方法である。一つの結晶から全ての参照データセットを測定するため、吸収の差による系統的な誤差が乗りづらく、精度の高い位相決定を行なえる。しかし、吸収端近傍での異常分散効果の変化を正確に測定しなければならないため、まず吸収端の測定を行なわなければならない。
吸収端はXAFS(X線吸収微細構造)実験を行うことにより決定する。PFの場合にはまず、GUI上で目的の原子を選択し、MCA測定(多重波高分析)を行なう。目的の原子が存在しているときには、図5のように二つのピークが得られる。図5だと、右が散乱X線、左が目的原子からの蛍光X線なので、左のピークだけを波長を変えながら測定していくと、蛍光スペクトルを得ることができる。図6のような蛍光スペクトルが得られれば、吸収端を容易に確認することができ、吸収端(edge)、最大ピーク(peak)の波長を決定できる。remote波長は吸収端から十分離れた任意の波長で、通常吸収端から数百eVだけ高エネルギー側の波長を利用することが多い。PFでは、原子散乱因子見積もりソフト(CHOOCH(6))を利用して自動的にこれらの波長を決定している(図7)。
測定波長が決まれば、上記回折強度データ収集と同様にデータセットを測定していく。通常は同じ条件で順次データセットを測定していくが、放射線損傷が大きい場合や露光時間が長く入射強度が変わってしまう場合には、inverse setting測定法を用いると良い。inverse setting測定法はバイフット対の領域を交互に測定する方法で、異常散乱効果によるバイフット対間の回折強度差を精度よく測定することができる。3波長以上で測定するため、典型的なMAD実験でも約30分の時間がかかる。また、一定角度で波長をシフトし(例えばpeak10度⇒edge10度⇒remote10度)、それを繰り返す測定法も、放射線損傷を受け易い結晶の際には効果的な場合がある。
単波長異常分散法(SAD法)は最も吸収効果が期待される波長でデータ測定を行い、バイフット対間の回折強度差を利用して位相決定する方法である。同様にXAFS実験から吸収端を決定し、peak近傍でのデータ測定を行なう。これもinverse setting測定法を用いると良い場合がある。
4’‘)軽原子を用いた低エネルギーSAD実験
天然蛋白質や核酸に含まれるイオウやリン、カリウム、カルシウムなどの軽原子の異常散乱効果を利用した位相決定法である。軽原子からの異常散乱シグナルは微弱であるため、以前は適用することが困難であったが、放射光の利用や測定装置の高度化、解析アルゴリズムの進歩等により、測定や解析の精度が向上し、適用範囲が広がってきている。何よりも天然に含まれる原子のシグナルのみを利用して位相決定できるため、蛋白質結晶構造解析のブレイクスルーとして現在最も注目を集めている手法である。
測定は通常6keV近辺(約2.0Å)の波長を用い、多重度が10以上程度になるように振動範囲を設定する。その他の測定方法は上記回折強度データ収集と同様である。振動範囲が大きいため、測定時間が増加する。また、通常の放射光ビームラインでは12keV近辺(約1.0Å)に最適化されているので、さらに時間がかかる場合が多い。ただし、最近増加している低エネルギー用のビームライン、例えばPFのBL-17Aでは、だいたい30分程度でデータ測定を完了することができる。
4’’‘)超高分解能回折データ測定
酵素反応ではプロトン移動が重要な機能を果たしている場合が多く、反応機構の解明には水素を含めた構造情報が必要になる。水素原子の位置決定には1.0Åを切る超高分解能回折データが必要であり、放射光の高輝度性と波長可変性を利用することで可能になる。 測定は、光学分解能を稼ぐため、なるべく短波長のX線(例えば0.7Å)を使用する。高角の反射は角度幅が広がるため、なるべく平行度の高い光学系(ダブルスリット光学系など)を採用する。高角まで測定できるようなるべく大きな面積の検出器を利用する。また、高角と低角の強度差が大きいので、ダイナミックレンジの大きい(できれば16ビット以上)検出器を用いた方が良い。高角と低角と露光時間を変えて測定し、後でマージする方法も有効である(高角用の長時間露光測定の際には、検出器の飽和を避けるために低角領域に減衰板を用いると良い)。
データ数が十分多くなるので、解析の際には束縛条件を外した精密化を適用することができる。束縛条件が無い場合には、低分解能データの精度も重要である。電子密度を正確に計算するためにも重要になるので、丁寧に測定する必要がある。
5)データ処理及び保存
測定した回折像は、データ処理ソフト(HKL2000やmosflmなど)を利用して、指数付け、積分、精密化、平均化などの処理を行い、データセットにする。ビームラインによっては解析ソフトも装備しており、その場で構造解析を進めることもできる。PFのビームラインでは位相決定用の各種プログラムが用意されており、ビームラインにおいてデータ収集後すぐに位相決定が可能であり、その結果に応じて回折実験計画を練り直すことも可能になっている。また、PFでは構造解析用のスペースを設け、全てのビームラインと高速ネットワークで結ばれているため、実験終了後もオンサイトで処理や解析を行なうことができる。測定したデータを研究室に持ち帰って処理する場合、HKL2000やmosflmなどの処理ソフトを入手して処理する必要がある。上記のHKL2000は有償ソフト、mosflmはアカデミックユーザーは無償でダウンロードして使用することができる。
測定データはビームラインのファイルサーバに保存されている。USBやIEEEなどのバックアップ用のポートが装備されているので、それぞれのユーザーのディスクにファイル転送して持ち帰る。データ量が膨大でバックアップに時間がかかるため、差分バックアップソフトを仕掛けておくと片付けが楽である。後は利用報告書を提出して実験完了となる。
工夫とコツ
ビームラインの選定
放射光ビームラインには、大きく分けて挿入光源の高輝度ビームラインと、偏向電磁石の汎用ビームラインがある。高輝度ビームラインも、大強度性を重視したハイスループットビームラインと輝度を活かした微小集光ビームラインとに分けられる。また、それとは別に、エネルギー適性による区別もある。どのビームラインにも得手不得手があるので、これらの特性を十分に理解した上でビームラインを選定することが実験の成否のカギとなる。例えばPFでは、BL-5AとNW12A(ハイスループットビームライン)、BL-17A(微小集光ビームライン、低エネルギー利用)、BL-6A(汎用ビームライン)と分けられる。主に結晶サイズによって使い分け、ラフに言えば100μmよりも大きい場合にはハイスループットビームライン、数十ミクロン程度以下になると微小集光ビームラインの方が有利になる。軽原子SAD実験はもちろん低エネルギービームラインを利用するべきである。また、汎用ビームラインでは安定なビームが利用できるので、十分な結晶性能があれば良好なデータを得ることができる。検出器の露光面サイズも重要なポイントであり、高分解能測定や長大格子の結晶解析の際には大面積検出器が必要になる。
試料交換ロボットの活用
近年放射光ビームラインでは試料交換ロボットの導入が進められている。一度セットすれば実験ハッチ内に入らなくても実験が進められるので、高速化と省力化が図れる。ドラッグデザインのように数多くの複合体結晶の回折データ測定を進める場合には特に威力を発揮する。また、結晶品質のスクリーニングでも非常に有効である。一般的に蛋白質結晶の品質を制御することが難しく、例え同じ結晶バッチでも品質が異なる場合が多々ある。より多くの結晶を検査することで高精度なデータ測定に結びつけることができる。その他、開発が進んでいる遠隔実験、全自動測定、放射光施設によっては既に実現している依頼測定などでは試料交換ロボットの利用は必須である。
アニーリング法
凍結結晶の場合、短時間昇温させてから凍結し直すアニーリング法が有効な場合がある。分解能やモザイク性の悪い結晶を捨てる前には一度試してみることをお勧めする。PFのビームラインにはアニーリングシャッターが装備されており、実験ハッチ内に入ることなく吹き付け低温装置の気流を遮断することができる。シャッターはCCDカメラで観察しながら操作し、凍結母液が溶け切る程度を目安とする。昇温させる時間は概ね母液の粘性に関係しており、粘性が高い場合にはより長時間気流を遮断する。
MAD法における注意点
吸収端の波長は原子の周辺構造によりシフトするため、MAD法を行なう際には面倒でも蛍光スペクトルで吸収端の確認を行なうべきである。また、そのシフトの影響により、MCA測定において想定した波長では蛍光シグナルが検出されない場合がある。その場合には、想定波長よりも少し高エネルギー側でMCA測定を試してみるとシグナルが得られることがある。
MCAのシグナルは原子含量に比例する。シグナルが得られない場合には十分な位相決定への寄与ができない場合が多いため、調製条件の再検討をするべきであろう。
セレンや水銀、鉛など砒素の吸収端より高エネルギー側の原子でMADを試みる場合、砒素の異常散乱シグナルが影響するので、カコジル酸の使用は避けるべきである。
MAD法ではバイフット対を独立反射として扱うので、対象の低い結晶では非対称範囲以上の領域を測定する必要があることを留意しておくこと。逆に、照射線量が増すことによる放射線損傷の影響が大きい。場合によっては分解能を犠牲にして照射線量を落とす必要がある。
順番は、異常散乱効果の大きいpeakまたはedgeから測定する。後でSAD法解析を試みる場合にも有利である。
MADデータを処理する場合、全ての波長のデータを同じ結晶方位で処理してやる必要がある。異常分散効果も大きく、データの品質は良いのに解析できない場合、結晶方位を取り違えている場合が多い。
SAD法は、MAD法と違い位相が一義的に決まらないこと、情報が少ないために位相精度が悪いこと等が欠点であるが、照射線量を抑えられるため、放射線損傷の影響を受け易い結晶では効果的な手法である。結晶は多かれ少なかれ放射線損傷の影響を受けるので、MAD法による解析がうまくいかない場合、peak波長のデータセットだけを使ったSAD法解析を行なうのは大いに試す価値がある。
軽原子SAD法における注意点
この手法の成否は、微弱な異常分散シグナルの最大化と測定精度の高精度化である。上記軽原子の吸収端は4keV以下(約3.0Å)のため、通常利用されている12keV近傍(約1.0Å)から低エネルギーにすればするほど異常分散シグナルは増大するが、測定精度の方は各種吸収の影響を受け、悪化してゆく。そのため、現在の測定装置のセッティングでは6keV近傍(約2.0Å)が最も有効であるとされている(7)。しかし、6keVのX線でも吸収の効果は大きいため、微小なシグナルを精度よく測定するためには測定回数を増やす必要がある。だいたい目安としては多重度10ぐらいになるように振動範囲を決めてデータセットを測定する。また、結晶のマウントもなるべく母液を少なくするなどバックグラウンドを低減する努力をする必要がある。もう一つのカギは結晶性である。分解能はもちろん、低いモザイク性、均質性など、結晶性が良く高品質なデータセットが測定できれば成功率は向上する。蛋白質結晶の場合、照射してみなければ品質がわからないことが多く、とにかく測定してみることが一番である。そのためには高速でスクリーニングする必要があろう。
放射線損傷問題
ビームラインの高輝度化が進み、凍結試料でも放射線損傷の問題が顕在化している。最新のビームラインでは、結晶回折の限界線量(Henderson’s limit(8))すらも超えてしまうため、限界線量内で効率的な測定条件を見積もるソフトウェアRADDOSE等が活用されている。ただ、結晶毎に放射線耐性は異なり、限界線量内でも損傷が生じてくるため、注意して測定を進める必要がある。放射線損傷を受け易い結晶などでは、例えば同型結晶を用いて最大許容放射線量を事前に見積もっておく、複数結晶を用いて測定を行なう、等の工夫が必要になってくる。
遠隔実験
放射光施設へのアクセスは一つの問題である。現在は実際にビームラインに来て実験することが多いが、結晶だけ送付して実験ができれば省力化が図れる。放射光施設では、試料交換ロボット、自動位置決め機能、遠隔制御ソフト等の開発を進めており、近い将来自動測定もしくは遠隔実験が実現されるであろう。現在でも、放射光施設によって依頼測定を受け付けているところがあるので、活用してみるのも一考である。PFでは、遠隔実験モニタリングシステムのテスト運用を開始している。例えば研究室から一人だけビームラインに来て実験をし、その進展状況を研究室からウェブ上でチェックすることができるので、大人数でビームラインに来る必要がなくなる。また、共同研究先と進展状況を見ながら実験を進めることができる等の活用の仕方も考えられる。
文献
- http://www.hamptonresearch.com/
- Hiraki, M. et al., AIP Conf. Proc., 879, 1924-1927 (2007)
- Murray, J. W. et al., J. Appl. Cryst., 37, 513-522 (2004)
- Otwinowski, Z. & Minor, W., Methods in Enzymology, Volume 276 (ed. Carter, C. W. & Sweet, R. M.) 307-326, Academic Press, New York (1997)
- Leslie A. G. W., CCP4 ESF-EACMB Newslett. Protein Crystallogr. 26 (1992)
- Evans, G. & Pettifer, R. F., J. Appl. Cryst., 34, 82-86 (2001)
- Mueller-Dieckmann, C. et al., Acta Cryst., D61, 1263-1272 (2005)
- Henderson, R., Proc. R. Soc. London, B241, 6-8 (2008)
-

図1: -
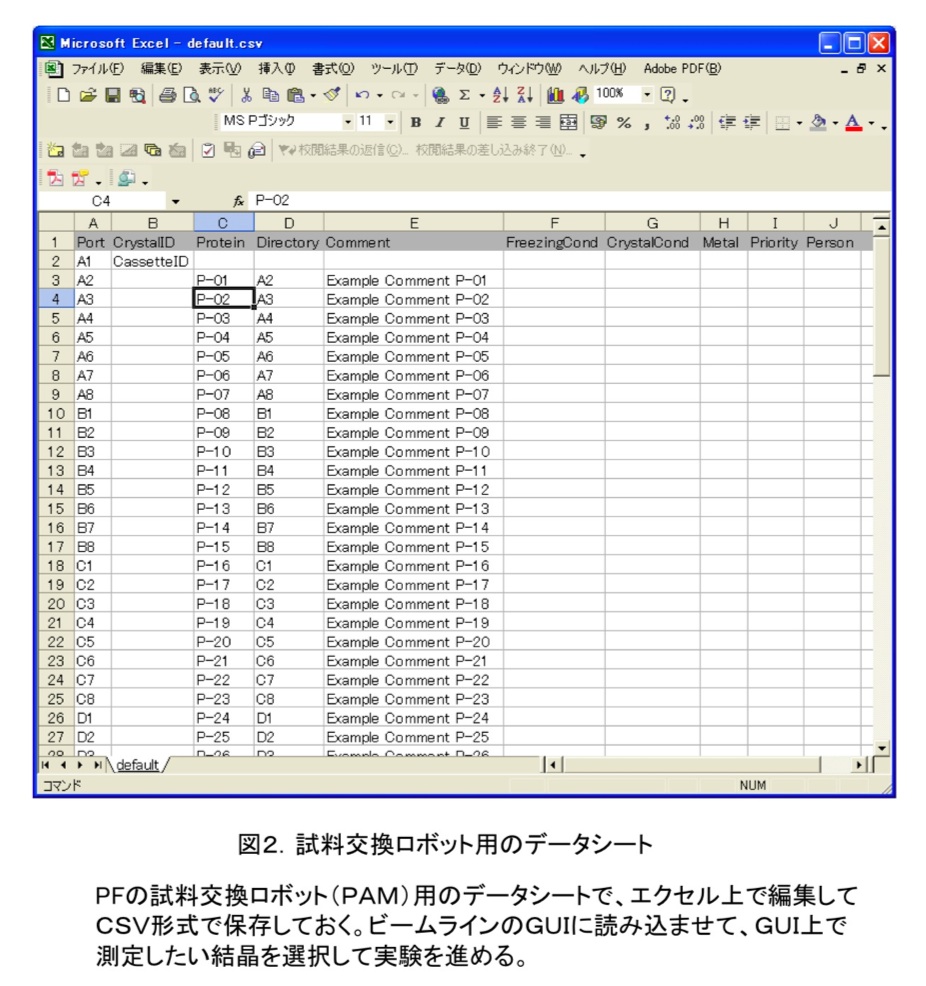
図2: -
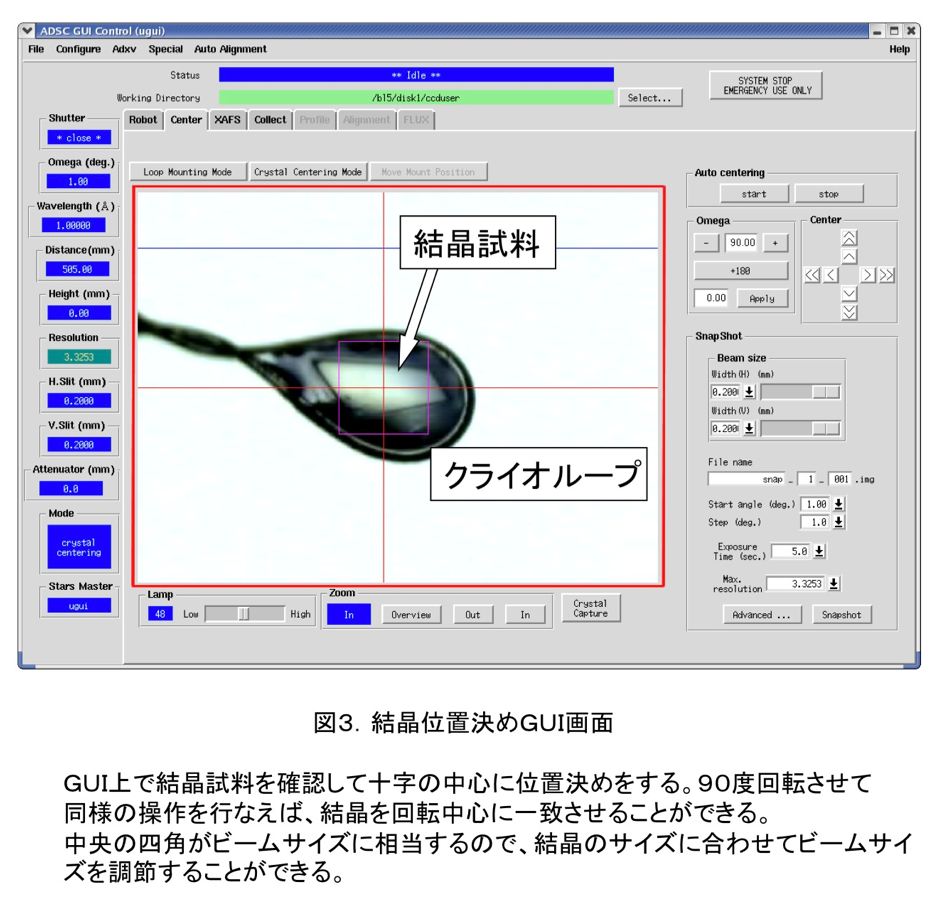
図3: -

図4: -
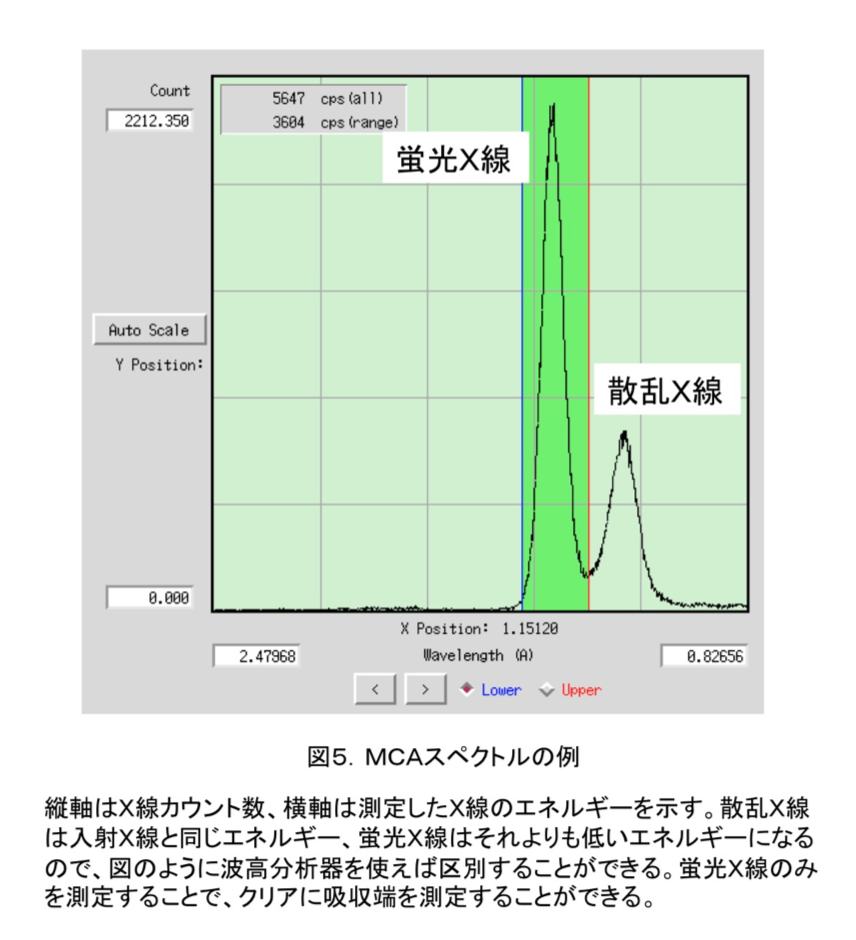
図5: -
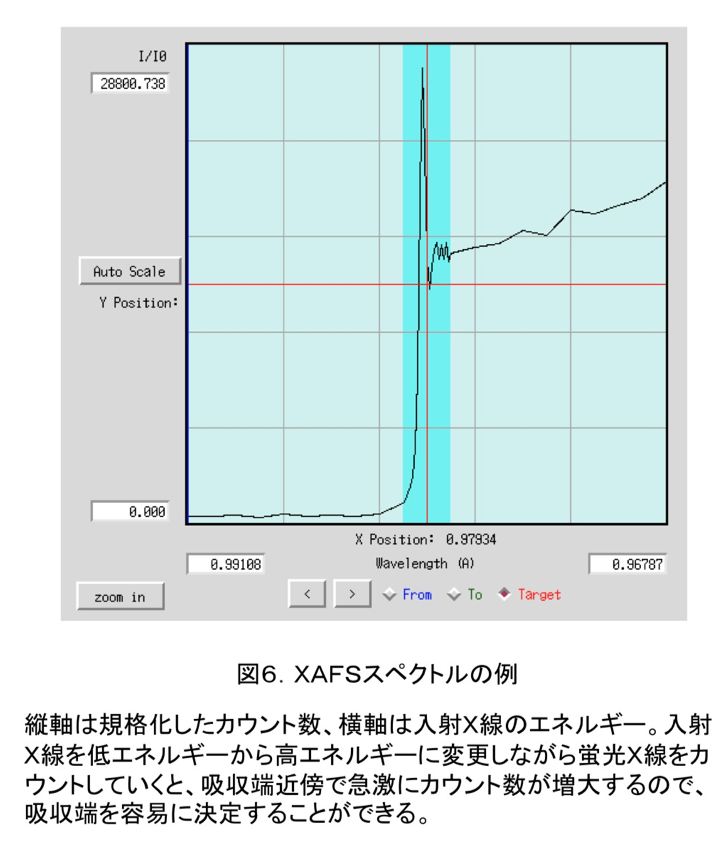
図6: -
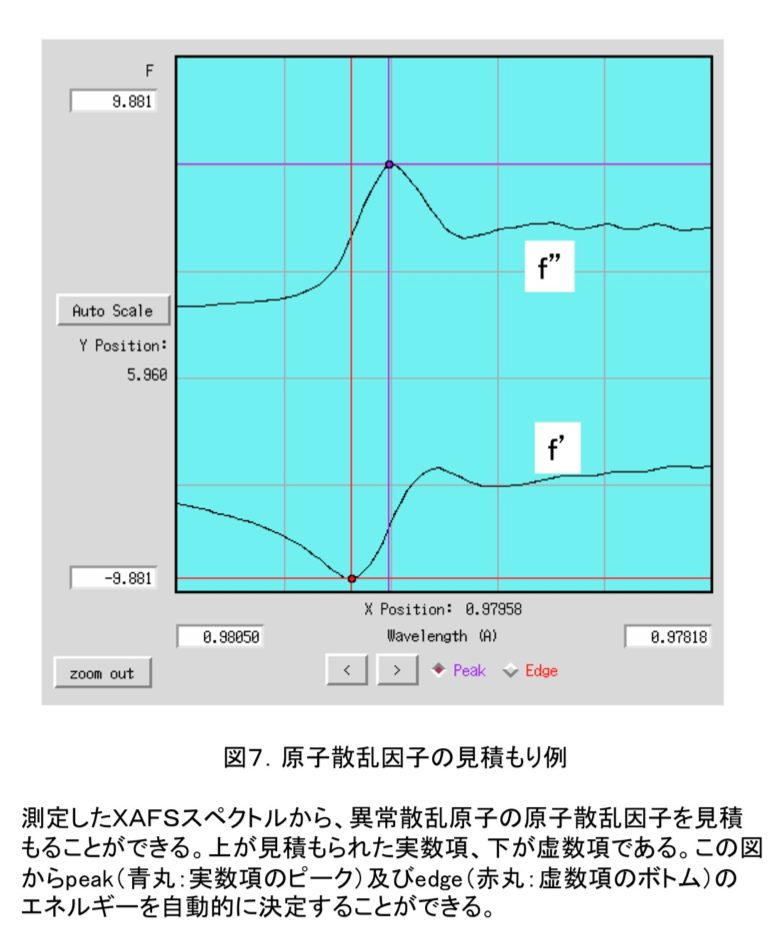
図7:
