

概要
PAタグシステムは一段階かつ非常に高い純度で目的蛋白質を精製できる新規アフィニティータグシステムである。さらに、PAタグはウェスタンブロッティングやフローサイトメトリー等の免疫検出においても使用可能である。本稿では、PAタグシステムを用いた蛋白質精製法およびPAタグを用いたウェスタンブロッティングについて、実験結果を交えながら解説する。また、PAタグを用いた精製や検出の操作は一日以内に十分可能である。なお、PAタグシステムは2014年3月現在ではまだ販売されていないが、今後市販される予定である。
目的・イントロダクション
蛋白質の機能を解析する上で純度が高い蛋白質を得ることは非常に重要である。現在蛋白質精製において、最もポピュラーな手法としてアフィニティータグシステムがある。なかでも「エピトープタグ」と総称される、ペプチドタグとそれに対するモノクローナル抗体からなるシステムは、FLAG、HA、Myc等の多くの種類のタグが開発され、販売されている。しかし、どのタグシステムも一長一短であり、欠点全てが克服されたようなタグシステムはなかなか存在しない。そこで私は、数々の欠点を克服した新規アフィニティータグシステム「PAタグシステム」を開発した。PAタグは、
NH2-GVAMPGAEDDVV-COOH
という12アミノ酸残基から成り、ラットモノクローナル抗体NZ-1と特異的に結合する。PAタグシステムは、現存するタグシステムと比較し、数々の利点を兼ね備えている。具体的には、12アミノ酸残基という短い配列、非常に高い親和性と特異性、目的蛋白質の溶出およびNZ-1を固定化したレジンの再生が容易に可能、等である。実際、私たちの研究室ではPAタグシステムを利用し、既に20種類以上の蛋白質の精製に成功している。一方、精製だけではなく検出にも使えることはタグの可能性を大きく広げ、より有用性を高めるが、PAタグは免疫学的手法によって目的の蛋白質のみを手早くかつ低コストで検出することも可能にする。セクションIでは、具体的な精製例も示しつつ、PAタグシステムを用いた蛋白質の精製法、特に哺乳類動物細胞発現系を用いた場合について解説する。そしてセクションIIではPAタグシステムを利用したウェスタンブロッティングでの抗原検出法について解説する。なお、本稿ではふれないが、PAタグ/NZ-1のシステムはフローサイトメトリーや免疫細胞化学的な検出(Immunocytochemistry : ICC)にも使用可能である(1, 2)。
装置・器具・試薬
- 遠心機(各社)
- インキュベーター(各社)
- 震盪装置(各社)
- ローテーター(各社)
- SDSゲル電気泳動装置(各社)
- Trans-Blot Turbo Transfer System (Bio-rad)
- ImageQuant LAS 4000mini(GE Healthcare)
- PEI(Sigma-Aldrich #408727)
- DMEM(各社)
- ウシ胎児血清(各社)
- CNBr-activated Sepharose 4FF(GE Healthcare)
- エコノカラム(Bio-rad)
- MES(各社)
- MgCl2(各社)
- Oriole染色キット(Bio-rad)
- HRP-conjugated rabbit anti-rat IgGポリクローナル抗体(Sigma-Aldrich)
- ECL Prime(GE Healthcare)
- PAタグペプチド(NH2-EGGVAMPGAEDDVV-COOH)(標準的なfmoc法やtBoc法で合成する。ペプチド受託合成サービスを利用すると良い。HPLCで純度95%以上に精製し、凍結乾燥したものは非常に良く水に溶ける。TFA塩のままで通常は特に差し支えない。)
実験手順
I 蛋白質精製
1)哺乳類動物細胞へのトランスフェクション
2)培養上清の回収および蛋白質のレジンへのキャプチャー
3)レジンの回収
4)レジンのwash
5)レジンからの溶出
6)レジンの再生
II ウェスタンブロッティング
1)SDS-PAGEおよびトランスファー
2)ブロッキング
3)一次抗体反応
4)二次抗体反応
5)検出
実験の詳細
I 蛋白質精製
基本的なトランスフェクション方法は、文献3のdishを用いた場合と同様である(3)。本稿ではアフィニティーカラムクロマトグラフィーのプロトコールはPAタグシステムに特化して記述している。また、PAタグシステムによる蛋白質精製の概略図を図1に示した。
1)哺乳類細胞へのトランスフェクション
本稿ではモデル蛋白質として、C末端にPAタグを付加させたmouse Neuropilin-1の細胞外ドメイン(mNRP1ec)の分泌型蛋白質を用いている。トランスフェクションは、15 cm dish(~25 ml medium)に約4-6割のHEK293T細胞へPEI(Sigma-Aldrich)を用いて行っている。そして、15 cm dish一枚に対し、20μgのDNAを使用し、その5倍量のPEI(100μg)を加えている。
2)培養上清の回収および蛋白質のレジンへのキャプチャー
トランスフェクションした培養細胞を37℃ 5% CO2条件下で約72時間インキュベーションする。その後、培養上清を回収し、終濃度が10 mMになるように1 M Tris-HCl(pH 8.0)を加え中和させる。そして、8000 rpmで20分以上遠心し、フィルターに通すことにより夾雑物を取り除く。そこに、NZ-1抗体をCNBr-activated Sepharose 4FFに約2 mg/mlになるように固定化したもの(NZ-1 Sepharose)をmedium量の1/100から1/200量加え、4℃で約2時間ゆっくり混和し、目的蛋白質をレジンへキャプチャーさせる。また、本稿ではバッチ法により精製を行っているが、カラム法による精製ももちろん可能である。
3)レジンの回収
目的蛋白質をキャプチャーさせたレジンを培養上清ごとデカンテーションでエコノカラム(Bio-rad)に移す。このとき、なるべくレジンの取りこぼしがないように、フロースルー液でレジン懸濁液が入っていた容器を何回か洗い込む。また、エコノカラムの径はレジン量や時間との兼ね合いで任意に決定する。
4)レジンのwash
カラムへの充填が終わったら、レジンの20倍量のwash bufferを流して洗浄する。本稿ではTBS(pH 7.5)[20 mM Tris-HCl,150 mM NaCl]でレジン量の4倍量×5回washを行っている。なおwash bufferには非常に低いpH(5.0以下)や、非常に高塩濃度(1 M NaClなど)のものでなければ、任意のbufferを使用可能である。
5)レジンからの溶出
washが完了したレジンから、0.1 mg/mlの競合ペプチド溶液(NH2-EGGVAMPGAEDDVV-COOH)あるいは10 mM MES・2 M MgCl2(pH 6.0)で溶出を行う。基本的にどちらの場合でも、レジンの10倍量の溶液で溶出を行えばキャプチャーされた蛋白質を9割以上溶出することが可能である。図2にはキャプチャーまでを同じ条件で行い、2種類の方法で溶出(レジン量の1倍量×10フラクション)した結果の比較を示す。なお競合ペプチド溶出の際には、elution bufferを一定量加えてレジン内に染み込ませた後、毎回5分以上インキュベーション(静置で良い)することが重要である。これはPAタグのNZ-1からの解離が遅いため、フリーのペプチドが競合するのに十分の時間を与えることが必要だからである。溶出法として競合ペプチドと高濃度マグネシウム溶液のどちらを選ぶかは、精製蛋白質のその後の使用用途や蛋白質自体への影響も考えて決めるとよい。
6)レジンの再生
使用済みのレジンは、10 mM MES・3 M MgCl2(pH 6.0)で再生することが可能である。レジン量に対し10倍量の上記再生液を加え、室温で10分以上ゆっくり混和する。この作業をbuffer交換しながら3回繰り返すことにより再生する。最後に、TBS等でよく洗浄してマグネシウムを取り除けば、次回以降も使用することが可能である。ただし、使用後のレジンからSDSで溶出されてくる蛋白質を電気泳動してみると(図3)、レジンから外れてくるNZ-1抗体のバンド以外に、再生液で溶出されずに結合したままだったタグ付き蛋白質のバンドがうっすらと見えてしまうことがあり、100%の溶出・再生はできていないことがわかる。再生したレジンは蛋白質精製に繰り返し使用可能であることは確認済みだが、異なる蛋白質の精製を同じレジンで行う時にはコンタミネーションの可能性について注意が必要である。
II ウェスタンブロッティング
ウェスタンブロッティングにおける手法や注意点は、文献4と同様である(4)。本稿ではPAタグを用いた手法に特化して記述していく。
1)SDS-PAGEおよびトランスファー
任意の量のサンプルを用意し、SDS-PAGEを行う。PAタグシステムは非常に高感度なので、サンプル量が多すぎると目的とするバンドが大きくなりすぎてしまい、そのバンド周辺については正確な情報が得られなくなってしまう。よって、正確なデータを出すためには、サンプル量を何回か検討し、最適なサンプル量を見極めることが必要である。本稿では,サンプルとしてosteosarcoma細胞に発現させたイソクエン酸脱水素酵素(IDH1)を用いた。このIDH1はC末端側にタンデムにFLAGタグとPAタグを付加させている(IDH1-FLAG-PA)。そして、1レーンあたり10μgの蛋白質(サンプル全体の蛋白質量)をSDS-PAGEした。SDS-PAGE後のゲルはなるべく手で触れずに転写膜(PVDFあるいはニトロセルロース)へとのせ、膜への転写を行う。本稿では、Trans-Blot Turbo Transfer System(Bio-rad)を用いてPVDF膜への転写を行った。もし、蛋白質がしっかりと膜へ転写できているかどうか不安な場合や、Stainedのマーカーを持っていなかった場合は、ポンソーSで転写膜を染色すれば膜に転写された蛋白質を全て確認することができる。
2)ブロッキング
基本的には4%スキムミルク/TBST(pH 8.0)[10 mM Tris-HCl,150 mM NaCl,0.05% Tween 20]あるいは4%スキムミルク/PBST[PBS(pH 7.4),0.05% Tween 20]中で、室温で15分間震盪することによってブロッキングを行う。もし、スキムミルクでのブロッキングが上手く行かなかったならば、4%スキムミルクの代わりに2% BSAを用いてもブロッキングは可能である。また、ブロッキング後は転写膜をTBSTに浸し、bufferを交換しながら室温で5分間×4回震盪することによって、転写膜をwashする。
3)一次抗体反応
一次抗体がNZ-1の場合では1μg/mlになるようにTBSTで希釈したもので濃度は十分である。そして、転写膜を一次抗体溶液に浸した状態で、室温で30分間震盪させることによって、NZ-1を転写膜上のPAタグ付き蛋白質に結合させる。なお、一次抗体溶液は回収し、4℃で保存しておけば、複数回使い回すことが可能である。また、一次抗体反応後はブロッキング後と同様に転写膜をwashする。
4)二次抗体反応
NZ-1はrat抗体であり、検出には一般的なペルオキシダーゼ(HRP)の酵素反応を用いるので、本稿では二次抗体にHRP-conjugated rabbit anti-rat IgGポリクローナル抗体(Sigma-Aldrich)を用いた。二次抗体もTBSTで希釈しているが、購入した二次抗体のロットによって感度は異なってくるため、各々で確立している濃度で使用すればよい。参考までに私たちの研究室では、1/6000希釈で用いている。そして、転写膜を二次抗体溶液に浸した状態で、室温で30分間震盪させることによって、二次抗体を転写膜上のNZ-1に結合させる。また、二次抗体反応後はブロッキング後と同様に転写膜をwashする。
5)検出
本稿ではHRP反応の検出試薬にECL Prime(GE Healthcare)を用いているが、他のHRP用検出試薬でも問題はない。また、発光の検出にはImageQuant LAS 4000mini(GE Healthcare)を用いている。本稿の条件では、基本的には10秒も露光すれば十分なバンドを確認することができる。ただし、これは検出試薬やイメージャーの感度、そして検出したい蛋白質の濃度に依存するので、適宜最適化を行う必要がある。
実際のウェスタンブロッティングの結果として、PAタグとFLAGタグのウェスタンブロッティングの結果および転写膜をそのままCBB染色した結果の比較の図をのせた(図4)。PAタグのウェスタンブロッティングの手法は前述通りだが、FLAGタグ抗体でのウェスタンブロッティングの方法は、一次抗体として3.5μg/ml の抗FLAGタグM2抗体(Sigma-Aldrich)、二次抗体に1/6000希釈HRP-conjugated goat anti-mouse IgG ポリクローナル抗体(Sigma-Aldrich)を用いている他は、上記のPAタグの場合と全く同様に行った。二次抗体が異なるため単純な比較はできないが、このPAタグによるウェスタンブロッティングのレーンと、FLAGタグによるウェスタンブロッティングのレーンと、転写膜自体をCBB染色し転写された蛋白質全てを可視化したレーンを比較すると、PAタグのレーンは非特異的なバンドがほとんど見えず、目的のバンドを明確に確認することができる。さらにPAタグ/NZ-1のシステムは、NZ-1の濃度を低く(0.01μg/ml)あるいはインキュベーション時間を短く(1 min)しても目的蛋白質のバンドが検出可能なことから(1)、ウェスタンブロッティングにおける感度は十分に高いことが確認できる。
工夫とコツ
溶出時のインキュベーション
溶出時に5分以上インキュベーションすることは非常に重要である。フラクションごとにインキュベーションをせずに連続的に蛋白質の溶出を行うと、シャープな溶出ができず、広い範囲にわたって低濃度の試料を含む溶出フラクションが出てきてしまう。そのため最終的な精製蛋白質が希釈され、同時に蛋白質収量の低下を導いてしまう。
MgCl2の取り扱い
高濃度マグネシウム溶液は、水酸化物イオンの存在下では難溶性の水酸化マグネシウム(Mg(OH)2)が形成されてしまうため、非常に沈殿しやすい。そのため、高濃度マグネシウム溶液を作製する際には、まずpHを合わせた高濃度MES溶液等を作製しておき、それにMilliQ水とMgCl2を加えることにより調整する。すると、NaOHを使わずともほぼ目的通りのpHに調整することができる。また、SDS-PAGEのサンプルを調製する際にも沈殿が生じてしまうので不便だが、この問題はサンプルをTBS等のbufferで1/10希釈してマグネシウム濃度を落としてからSDS-PAGEし、銀染色や蛍光色素染色などの高感度法でバンドを可視化することや、TCA沈殿などで脱塩してから泳動することで避けられる。また、2 M MgCl2を用いて溶出したサンプルを透析でbuffer交換する際には、体積が2倍程度まで増えてしまうので、透析チューブはその分を見越して長めに切って使用する必要がある。
吸光度計を用いた精製蛋白質の定量
PAタグペプチドの配列はNH2-EGGVAMPGAEDDVV-COOHであるため芳香族アミノ酸を含んでいない。よって、SDS-PAGEを行う前に、各溶出フラクションのA280を測定することによって、どのフラクションに蛋白質が多く溶出されているのか簡便に見積もることが可能である。
PEIによるトランスフェクション
PEIには細胞毒性があるが販売会社ごとにその特性は異なっている。そのため、実際にトランスフェクションを行う場合には、商品ごとに条件検討を行う必要がある。参考までに私たちは、Sigma-Aldrich社製のPEI(#408727)を用いてDNA:PEI=1:5になるように調整し、トランスフェクションを行っている。
NZ-1のサブクラス
NZ-1はrat IgG2a lambdaに分類される。そして、一般的なこのサブクラスの抗体と同様に、Protein Gとは結合するが、Protein Aとは結合しにくく、kappa鎖に対して結合するProtein Lには結合しない。
ポドプラニン発現細胞株
元々NZ-1はヒトポドプラニンに対する抗体である(5)。そして、HEK293T、COS-1、COS-7細胞にはポドプラニンが発現しており、NZ-1と反応してしまう。そのため、これらの細胞株のlysateをウェスタンブロッティングすると37 kDa近辺にポドプラニンのバンドが見えてしまう。さらに、ポドプラニンは細胞表面に発現しているため、これらの細胞株はフローサイトメトリーを用いたPAタグ付加受容体などの解析には使用することができない。しかし、ポドプラニンはmedium中には全く分泌あるいはシェディングされないので、分泌型の蛋白質の発現精製には問題なくこれらの細胞株でも使用可能である。また、HEK細胞で発現した膜蛋白質の精製を行うと、不思議なことに精製試料の中にポドプラニンは混入してこない(1)。ポドプラニンは糖蛋白質であり、PAタグ配列の直後にO型糖鎖が付加されることが知られているので、細胞上に発現したポドプラニンに対するNZ-1の親和性はPAタグに対するそれよりかなり低いのかもしれない。ちなみに、ポドプラニン中のPAタグに相当する部分の配列はヒト(サルと同一)とそれ以外の動物(マウス、ラット、ハムスターなど)で大きく異なっており、NZ-1はこれらの動物種由来の細胞には全く反応しない。
文献
- Fujii Y. et al., Protein Expr. Purif. 95, 240-7 (2014)
- Kato-Kaneko M. et al., Biochem. Biophys. Res. Commun. 432, 40-5 (2013)
- 橋口隆生ら, 蛋白質科学会アーカイブ, 1, e017 (2008)
- 恩田真紀, 蛋白質科学会アーカイブ, 1, e012 (2008)
- Kato Y. et al., Biochem. Biophys. Res. Commun. 349, 1301-7 (2006)
-
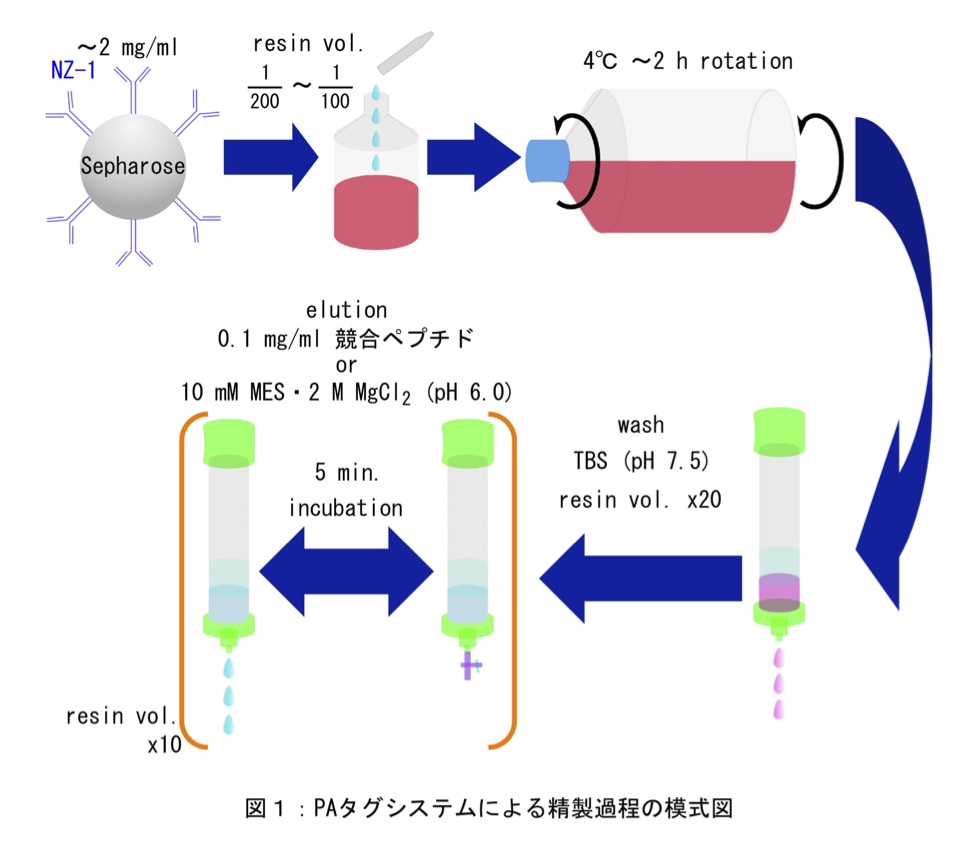
図1 -

図2 -

図3 -
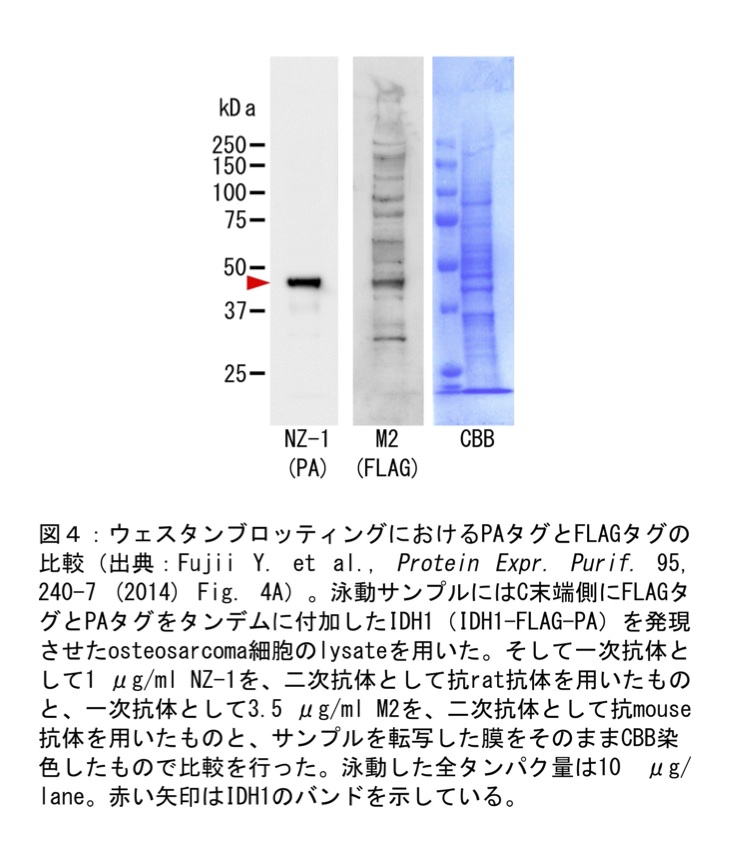
図4
概要
PAタグシステムは一段階かつ非常に高い純度で目的蛋白質を精製できる新規アフィニティータグシステムである。さらに、PAタグはウェスタンブロッティングやフローサイトメトリー等の免疫検出においても使用可能である。本稿では、PAタグシステムを用いた蛋白質精製法およびPAタグを用いたウェスタンブロッティングについて、実験結果を交えながら解説する。また、PAタグを用いた精製や検出の操作は一日以内に十分可能である。なお、PAタグシステムは2014年3月現在ではまだ販売されていないが、今後市販される予定である。
目的・イントロダクション
蛋白質の機能を解析する上で純度が高い蛋白質を得ることは非常に重要である。現在蛋白質精製において、最もポピュラーな手法としてアフィニティータグシステムがある。なかでも「エピトープタグ」と総称される、ペプチドタグとそれに対するモノクローナル抗体からなるシステムは、FLAG、HA、Myc等の多くの種類のタグが開発され、販売されている。しかし、どのタグシステムも一長一短であり、欠点全てが克服されたようなタグシステムはなかなか存在しない。そこで私は、数々の欠点を克服した新規アフィニティータグシステム「PAタグシステム」を開発した。PAタグは、
NH2-GVAMPGAEDDVV-COOH
という12アミノ酸残基から成り、ラットモノクローナル抗体NZ-1と特異的に結合する。PAタグシステムは、現存するタグシステムと比較し、数々の利点を兼ね備えている。具体的には、12アミノ酸残基という短い配列、非常に高い親和性と特異性、目的蛋白質の溶出およびNZ-1を固定化したレジンの再生が容易に可能、等である。実際、私たちの研究室ではPAタグシステムを利用し、既に20種類以上の蛋白質の精製に成功している。一方、精製だけではなく検出にも使えることはタグの可能性を大きく広げ、より有用性を高めるが、PAタグは免疫学的手法によって目的の蛋白質のみを手早くかつ低コストで検出することも可能にする。セクションIでは、具体的な精製例も示しつつ、PAタグシステムを用いた蛋白質の精製法、特に哺乳類動物細胞発現系を用いた場合について解説する。そしてセクションIIではPAタグシステムを利用したウェスタンブロッティングでの抗原検出法について解説する。なお、本稿ではふれないが、PAタグ/NZ-1のシステムはフローサイトメトリーや免疫細胞化学的な検出(Immunocytochemistry : ICC)にも使用可能である(1, 2)。
装置・器具・試薬
- 遠心機(各社)
- インキュベーター(各社)
- 震盪装置(各社)
- ローテーター(各社)
- SDSゲル電気泳動装置(各社)
- Trans-Blot Turbo Transfer System (Bio-rad)
- ImageQuant LAS 4000mini(GE Healthcare)
- PEI(Sigma-Aldrich #408727)
- DMEM(各社)
- ウシ胎児血清(各社)
- CNBr-activated Sepharose 4FF(GE Healthcare)
- エコノカラム(Bio-rad)
- MES(各社)
- MgCl2(各社)
- Oriole染色キット(Bio-rad)
- HRP-conjugated rabbit anti-rat IgGポリクローナル抗体(Sigma-Aldrich)
- ECL Prime(GE Healthcare)
- PAタグペプチド(NH2-EGGVAMPGAEDDVV-COOH)(標準的なfmoc法やtBoc法で合成する。ペプチド受託合成サービスを利用すると良い。HPLCで純度95%以上に精製し、凍結乾燥したものは非常に良く水に溶ける。TFA塩のままで通常は特に差し支えない。)
実験手順
I 蛋白質精製
1)哺乳類動物細胞へのトランスフェクション
2)培養上清の回収および蛋白質のレジンへのキャプチャー
3)レジンの回収
4)レジンのwash
5)レジンからの溶出
6)レジンの再生
II ウェスタンブロッティング
1)SDS-PAGEおよびトランスファー
2)ブロッキング
3)一次抗体反応
4)二次抗体反応
5)検出
実験の詳細
I 蛋白質精製
基本的なトランスフェクション方法は、文献3のdishを用いた場合と同様である(3)。本稿ではアフィニティーカラムクロマトグラフィーのプロトコールはPAタグシステムに特化して記述している。また、PAタグシステムによる蛋白質精製の概略図を図1に示した。
1)哺乳類細胞へのトランスフェクション
本稿ではモデル蛋白質として、C末端にPAタグを付加させたmouse Neuropilin-1の細胞外ドメイン(mNRP1ec)の分泌型蛋白質を用いている。トランスフェクションは、15 cm dish(~25 ml medium)に約4-6割のHEK293T細胞へPEI(Sigma-Aldrich)を用いて行っている。そして、15 cm dish一枚に対し、20μgのDNAを使用し、その5倍量のPEI(100μg)を加えている。
2)培養上清の回収および蛋白質のレジンへのキャプチャー
トランスフェクションした培養細胞を37℃ 5% CO2条件下で約72時間インキュベーションする。その後、培養上清を回収し、終濃度が10 mMになるように1 M Tris-HCl(pH 8.0)を加え中和させる。そして、8000 rpmで20分以上遠心し、フィルターに通すことにより夾雑物を取り除く。そこに、NZ-1抗体をCNBr-activated Sepharose 4FFに約2 mg/mlになるように固定化したもの(NZ-1 Sepharose)をmedium量の1/100から1/200量加え、4℃で約2時間ゆっくり混和し、目的蛋白質をレジンへキャプチャーさせる。また、本稿ではバッチ法により精製を行っているが、カラム法による精製ももちろん可能である。
3)レジンの回収
目的蛋白質をキャプチャーさせたレジンを培養上清ごとデカンテーションでエコノカラム(Bio-rad)に移す。このとき、なるべくレジンの取りこぼしがないように、フロースルー液でレジン懸濁液が入っていた容器を何回か洗い込む。また、エコノカラムの径はレジン量や時間との兼ね合いで任意に決定する。
4)レジンのwash
カラムへの充填が終わったら、レジンの20倍量のwash bufferを流して洗浄する。本稿ではTBS(pH 7.5)[20 mM Tris-HCl,150 mM NaCl]でレジン量の4倍量×5回washを行っている。なおwash bufferには非常に低いpH(5.0以下)や、非常に高塩濃度(1 M NaClなど)のものでなければ、任意のbufferを使用可能である。
5)レジンからの溶出
washが完了したレジンから、0.1 mg/mlの競合ペプチド溶液(NH2-EGGVAMPGAEDDVV-COOH)あるいは10 mM MES・2 M MgCl2(pH 6.0)で溶出を行う。基本的にどちらの場合でも、レジンの10倍量の溶液で溶出を行えばキャプチャーされた蛋白質を9割以上溶出することが可能である。図2にはキャプチャーまでを同じ条件で行い、2種類の方法で溶出(レジン量の1倍量×10フラクション)した結果の比較を示す。なお競合ペプチド溶出の際には、elution bufferを一定量加えてレジン内に染み込ませた後、毎回5分以上インキュベーション(静置で良い)することが重要である。これはPAタグのNZ-1からの解離が遅いため、フリーのペプチドが競合するのに十分の時間を与えることが必要だからである。溶出法として競合ペプチドと高濃度マグネシウム溶液のどちらを選ぶかは、精製蛋白質のその後の使用用途や蛋白質自体への影響も考えて決めるとよい。
6)レジンの再生
使用済みのレジンは、10 mM MES・3 M MgCl2(pH 6.0)で再生することが可能である。レジン量に対し10倍量の上記再生液を加え、室温で10分以上ゆっくり混和する。この作業をbuffer交換しながら3回繰り返すことにより再生する。最後に、TBS等でよく洗浄してマグネシウムを取り除けば、次回以降も使用することが可能である。ただし、使用後のレジンからSDSで溶出されてくる蛋白質を電気泳動してみると(図3)、レジンから外れてくるNZ-1抗体のバンド以外に、再生液で溶出されずに結合したままだったタグ付き蛋白質のバンドがうっすらと見えてしまうことがあり、100%の溶出・再生はできていないことがわかる。再生したレジンは蛋白質精製に繰り返し使用可能であることは確認済みだが、異なる蛋白質の精製を同じレジンで行う時にはコンタミネーションの可能性について注意が必要である。
II ウェスタンブロッティング
ウェスタンブロッティングにおける手法や注意点は、文献4と同様である(4)。本稿ではPAタグを用いた手法に特化して記述していく。
1)SDS-PAGEおよびトランスファー
任意の量のサンプルを用意し、SDS-PAGEを行う。PAタグシステムは非常に高感度なので、サンプル量が多すぎると目的とするバンドが大きくなりすぎてしまい、そのバンド周辺については正確な情報が得られなくなってしまう。よって、正確なデータを出すためには、サンプル量を何回か検討し、最適なサンプル量を見極めることが必要である。本稿では,サンプルとしてosteosarcoma細胞に発現させたイソクエン酸脱水素酵素(IDH1)を用いた。このIDH1はC末端側にタンデムにFLAGタグとPAタグを付加させている(IDH1-FLAG-PA)。そして、1レーンあたり10μgの蛋白質(サンプル全体の蛋白質量)をSDS-PAGEした。SDS-PAGE後のゲルはなるべく手で触れずに転写膜(PVDFあるいはニトロセルロース)へとのせ、膜への転写を行う。本稿では、Trans-Blot Turbo Transfer System(Bio-rad)を用いてPVDF膜への転写を行った。もし、蛋白質がしっかりと膜へ転写できているかどうか不安な場合や、Stainedのマーカーを持っていなかった場合は、ポンソーSで転写膜を染色すれば膜に転写された蛋白質を全て確認することができる。
2)ブロッキング
基本的には4%スキムミルク/TBST(pH 8.0)[10 mM Tris-HCl,150 mM NaCl,0.05% Tween 20]あるいは4%スキムミルク/PBST[PBS(pH 7.4),0.05% Tween 20]中で、室温で15分間震盪することによってブロッキングを行う。もし、スキムミルクでのブロッキングが上手く行かなかったならば、4%スキムミルクの代わりに2% BSAを用いてもブロッキングは可能である。また、ブロッキング後は転写膜をTBSTに浸し、bufferを交換しながら室温で5分間×4回震盪することによって、転写膜をwashする。
3)一次抗体反応
一次抗体がNZ-1の場合では1μg/mlになるようにTBSTで希釈したもので濃度は十分である。そして、転写膜を一次抗体溶液に浸した状態で、室温で30分間震盪させることによって、NZ-1を転写膜上のPAタグ付き蛋白質に結合させる。なお、一次抗体溶液は回収し、4℃で保存しておけば、複数回使い回すことが可能である。また、一次抗体反応後はブロッキング後と同様に転写膜をwashする。
4)二次抗体反応
NZ-1はrat抗体であり、検出には一般的なペルオキシダーゼ(HRP)の酵素反応を用いるので、本稿では二次抗体にHRP-conjugated rabbit anti-rat IgGポリクローナル抗体(Sigma-Aldrich)を用いた。二次抗体もTBSTで希釈しているが、購入した二次抗体のロットによって感度は異なってくるため、各々で確立している濃度で使用すればよい。参考までに私たちの研究室では、1/6000希釈で用いている。そして、転写膜を二次抗体溶液に浸した状態で、室温で30分間震盪させることによって、二次抗体を転写膜上のNZ-1に結合させる。また、二次抗体反応後はブロッキング後と同様に転写膜をwashする。
5)検出
本稿ではHRP反応の検出試薬にECL Prime(GE Healthcare)を用いているが、他のHRP用検出試薬でも問題はない。また、発光の検出にはImageQuant LAS 4000mini(GE Healthcare)を用いている。本稿の条件では、基本的には10秒も露光すれば十分なバンドを確認することができる。ただし、これは検出試薬やイメージャーの感度、そして検出したい蛋白質の濃度に依存するので、適宜最適化を行う必要がある。
実際のウェスタンブロッティングの結果として、PAタグとFLAGタグのウェスタンブロッティングの結果および転写膜をそのままCBB染色した結果の比較の図をのせた(図4)。PAタグのウェスタンブロッティングの手法は前述通りだが、FLAGタグ抗体でのウェスタンブロッティングの方法は、一次抗体として3.5μg/ml の抗FLAGタグM2抗体(Sigma-Aldrich)、二次抗体に1/6000希釈HRP-conjugated goat anti-mouse IgG ポリクローナル抗体(Sigma-Aldrich)を用いている他は、上記のPAタグの場合と全く同様に行った。二次抗体が異なるため単純な比較はできないが、このPAタグによるウェスタンブロッティングのレーンと、FLAGタグによるウェスタンブロッティングのレーンと、転写膜自体をCBB染色し転写された蛋白質全てを可視化したレーンを比較すると、PAタグのレーンは非特異的なバンドがほとんど見えず、目的のバンドを明確に確認することができる。さらにPAタグ/NZ-1のシステムは、NZ-1の濃度を低く(0.01μg/ml)あるいはインキュベーション時間を短く(1 min)しても目的蛋白質のバンドが検出可能なことから(1)、ウェスタンブロッティングにおける感度は十分に高いことが確認できる。
工夫とコツ
溶出時のインキュベーション
溶出時に5分以上インキュベーションすることは非常に重要である。フラクションごとにインキュベーションをせずに連続的に蛋白質の溶出を行うと、シャープな溶出ができず、広い範囲にわたって低濃度の試料を含む溶出フラクションが出てきてしまう。そのため最終的な精製蛋白質が希釈され、同時に蛋白質収量の低下を導いてしまう。
MgCl2の取り扱い
高濃度マグネシウム溶液は、水酸化物イオンの存在下では難溶性の水酸化マグネシウム(Mg(OH)2)が形成されてしまうため、非常に沈殿しやすい。そのため、高濃度マグネシウム溶液を作製する際には、まずpHを合わせた高濃度MES溶液等を作製しておき、それにMilliQ水とMgCl2を加えることにより調整する。すると、NaOHを使わずともほぼ目的通りのpHに調整することができる。また、SDS-PAGEのサンプルを調製する際にも沈殿が生じてしまうので不便だが、この問題はサンプルをTBS等のbufferで1/10希釈してマグネシウム濃度を落としてからSDS-PAGEし、銀染色や蛍光色素染色などの高感度法でバンドを可視化することや、TCA沈殿などで脱塩してから泳動することで避けられる。また、2 M MgCl2を用いて溶出したサンプルを透析でbuffer交換する際には、体積が2倍程度まで増えてしまうので、透析チューブはその分を見越して長めに切って使用する必要がある。
吸光度計を用いた精製蛋白質の定量
PAタグペプチドの配列はNH2-EGGVAMPGAEDDVV-COOHであるため芳香族アミノ酸を含んでいない。よって、SDS-PAGEを行う前に、各溶出フラクションのA280を測定することによって、どのフラクションに蛋白質が多く溶出されているのか簡便に見積もることが可能である。
PEIによるトランスフェクション
PEIには細胞毒性があるが販売会社ごとにその特性は異なっている。そのため、実際にトランスフェクションを行う場合には、商品ごとに条件検討を行う必要がある。参考までに私たちは、Sigma-Aldrich社製のPEI(#408727)を用いてDNA:PEI=1:5になるように調整し、トランスフェクションを行っている。
NZ-1のサブクラス
NZ-1はrat IgG2a lambdaに分類される。そして、一般的なこのサブクラスの抗体と同様に、Protein Gとは結合するが、Protein Aとは結合しにくく、kappa鎖に対して結合するProtein Lには結合しない。
ポドプラニン発現細胞株
元々NZ-1はヒトポドプラニンに対する抗体である(5)。そして、HEK293T、COS-1、COS-7細胞にはポドプラニンが発現しており、NZ-1と反応してしまう。そのため、これらの細胞株のlysateをウェスタンブロッティングすると37 kDa近辺にポドプラニンのバンドが見えてしまう。さらに、ポドプラニンは細胞表面に発現しているため、これらの細胞株はフローサイトメトリーを用いたPAタグ付加受容体などの解析には使用することができない。しかし、ポドプラニンはmedium中には全く分泌あるいはシェディングされないので、分泌型の蛋白質の発現精製には問題なくこれらの細胞株でも使用可能である。また、HEK細胞で発現した膜蛋白質の精製を行うと、不思議なことに精製試料の中にポドプラニンは混入してこない(1)。ポドプラニンは糖蛋白質であり、PAタグ配列の直後にO型糖鎖が付加されることが知られているので、細胞上に発現したポドプラニンに対するNZ-1の親和性はPAタグに対するそれよりかなり低いのかもしれない。ちなみに、ポドプラニン中のPAタグに相当する部分の配列はヒト(サルと同一)とそれ以外の動物(マウス、ラット、ハムスターなど)で大きく異なっており、NZ-1はこれらの動物種由来の細胞には全く反応しない。
文献
- Fujii Y. et al., Protein Expr. Purif. 95, 240-7 (2014)
- Kato-Kaneko M. et al., Biochem. Biophys. Res. Commun. 432, 40-5 (2013)
- 橋口隆生ら, 蛋白質科学会アーカイブ, 1, e017 (2008)
- 恩田真紀, 蛋白質科学会アーカイブ, 1, e012 (2008)
- Kato Y. et al., Biochem. Biophys. Res. Commun. 349, 1301-7 (2006)
