

概要
ポリアクリルアミドゲル電気泳動(PAGE:Poly Acrylamide Gel Electrophoresis)は、分子量の概算や精製度のチェック等、蛋白質科学の現場で最も頻繁に利用される実験手法の一つである。高価な機器を必要とせず操作も簡便で、工夫次第で分子の立体構造や電荷の状態など非常に多くの情報が得られる。その手法・用途は多岐にわたるが、本稿では、基本中の基本である蛋白質をターゲットとした一次元のSDS-PAGEとNative-PAGEについてまず解説する。次に、蛋白質の安定性や電荷を評価できる尿素ゲル-PAGEと、ゲルの染色法について紹介する。頻繁に実施する手法ゆえ、「手早く、手軽に、気楽に」をモットーに、個々の研究室の事情に合ったシステムを構築する事が重要である。
主な内容
- SDS-PAGE(1)
- Tricine-SDS-PAGE(低分子蛋白質用SDS-PAGE)(2)
- Native-PAGE(3)
- 尿素-PAGE(4)
- CBB染色
- 銀染色
各PAGEの特徴
蛋白質分子のゲル内移動速度と「分子量」「形(コンフォメーション)」「電荷」の関係
- Native-PAGE:移動速度は「分子量」「形」「電荷」すべてに依属。
- SDS-PAGE:移動速度は「分子量」に依属。SDS変性により「形」「電荷」が無視できる。
- 尿素-PAGE:移動速度は「分子量」「電荷」に依属。尿素変性により「形」が無視できる。
- BN-PAGE:移動速度は「分子量」「形」に依属。CBBの結合により「電荷」が無視できる。
■ SDS-PAGE ■ ミニ60分
負電荷を持つ界面活性剤SDSが結合することで蛋白質が変性する。結果、ゲル内移動速度の三大要素:「分子量」「形」「電荷」の内、「形」と「電荷」が無視できるようになり、移動速度が「分子量」依属となる [*1]。よって、蛋白質の精製度チェックや分子量測定などに利用できる。
装置・器具
- 電気泳動槽と専用電源(各社)
- 泳動プレート、パッキン、コウムなどを含むセット一式(各社)
試薬
- 1.5 M Tris-HCl, pH 8.8 [*2]
- 1 M Tris-HCl, pH 6.8 [*2]
- 30%アクリルアミド-0.8%ビスアクリルアミド:遮光冷蔵保存 [*2]
- 10% SDS(sodium dodecyl sulfate)
- 10% APS(ammonium persulfate):用時調製 [*3]
- TEMED(N,N,N’,N’-tetramethyl ethylenediamine)
- 50% 2-プロパノール
- 泳動buffer:188 gグリシン+30.2 g Tris+10 g SDS+純水 →10 Lにする [*4]
- 4× loading buffer:40%グリセロール-8%SDS-0.4%BPB-0.2 M Tris-HCl, pH 6.8 [*5]
- 2-メルカプトエタノール
【Tips】
- *1
- 厳密には、ゲル濃度0の時、理論上ポリペプチドの移動速度は分子量とは無関に等しくなる(1)。
- *2
- これらの試薬とゲル調製に使う純水は0.45 μmのフィルターで濾過する。
- *3
- 濃度が10%であれば、使用後速やかに冷蔵保存すれば1、2日は使える。
- *4
- ★重要★Trisはシグマ社製T1503を使用。pHは調整しない。蛇口付きポリタンクに保存し、必要量を泳動槽に注げるようにしておくと便利。
- *5
- 調製時は加温してSDSを溶解させ、エッペンチューブなどに小分けして室温で保存する。室温下ではSDSが析出してくるので、使用前にエッペンチューブごと40℃程度の湯に浮かべて少し温める。
実験の流れ
泳動プレートの組み立て
↓
ゲル溶液の調製(分離ゲル、濃縮ゲルの2種類)[5分]
↓
分離ゲルの重合(固めること)[20分]
↓
泳動プレートの洗浄 [1分]
↓
濃縮ゲルの重合 [25分]
↓
レーンの洗浄 [1分]
↓
蛋白質サンプルをゲルにのせる
↓
泳動 [ミニゲルで60分]
↓
バンドの確認(固定・染色・脱色)[市販キット使用の場合30分]
実験手順の詳細
I. SDSゲルの作成
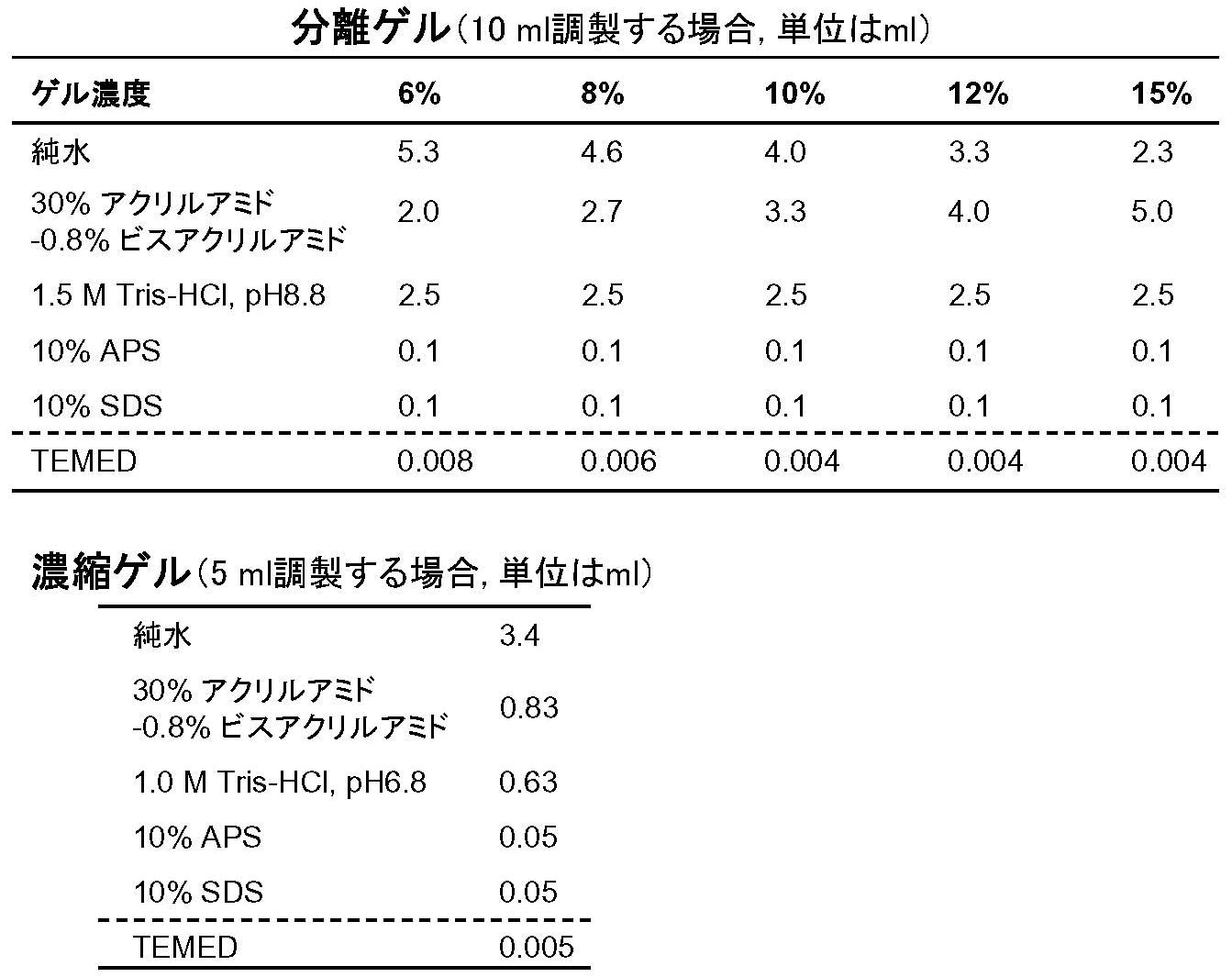
- 泳動プレートに、分離ゲル液を入れる目安となる線を油性ペンで引く [図1]。
- 泳動プレートをメーカーのマニュアルに従って組み立てる。
- [表1] に従ってTEMED以外の試薬を小ビーカー内で混合し、分離・濃縮ゲル溶液それぞれを調製する。溶液を泡立てないように細心の注意を払うこと。[*6]
- ビーカーに軽くラップでフタをして(密閉は×)デシケーターに入れ、真空ポンプ等を使って減圧脱気する。[省略可 *7]
- [表1] に従って分離ゲル溶液に重合反応開始剤TEMEDを加える。ゲルは直ぐに固まり始めるので、TEMED添加以降の作業は迅速に行うこと。[*8]
- 分離ゲル溶液をビーカーから直接泳動プレートの隙間に注ぐ。[*9]
- スポイトを使い、速やかに50% 2-プロパノールをぎりぎりいっぱいまで重層する。[*10]
- 20分間室温で放置しゲルを重合させる。
- プレートを逆さまにして2-プロパノールを捨て、洗瓶に入れた純水で分離ゲルの上側を洗浄する。洗浄後、プレートを逆さまにして振り、水分を切る。
- [表1] に従って濃縮ゲル溶液にTEMEDを加え、ビーカーから直接泳動プレートの間に注ぐ。差し込むコウムの体積を考慮し、注ぐゲルの量は切れ込みより少し手前にしておく [図1]。[*11]
- コウムを差込み、25分間室温で放置してゲルを重合させる。
- ★重要★コウムを抜き、直ぐに洗瓶に入れた純水でレーンを洗浄する。[*12]
- 泳動水槽にセットする。→「III. 電気泳動」へ
すぐに使わない場合は、キムワイプで包み、純水でキムワイプを十分湿らせてから、チャック付ビニル袋に入れて冷蔵保存する。ゲルの種類、調製日、調製者、レーンの数などを明記しておくと便利。
【Tips】
- *6
- ビーカーをゆっくり回転させて試薬を混合する。ガラス棒などで攪拌しない。特にSDSは泡立ち易いので、最後に静かに加え混合すること。調製容器はビーカーがベスト。
- *7
- ゲル溶液から気泡を抜く操作で、結果に差がなければ省略可。常圧に戻す際は一気に空気がデシケーター内に入らないよう注意すること。
- *8
- 室温が高い夏季には、氷上で操作を行うと重合速度を遅くすることができる。
- *9
- スポイト等を使用して注がない(気泡が入る)。
- *10
- 水、水飽和ブタノールを使用することもあるが、筆者のお勧めは50% 2-プロパノールである。また、なるべく多く重層した方がゲル界面の仕上がりが良い。
- *11
- 注ぐゲルが少な過ぎるとレーンが浅くなりサンプルをのせ難くなる。筆者の場合、コウムを差した時に少しゲルが溢れ出てくる程度まで入れている。
- *12
- 未重合のゲル溶液を除く。この操作を怠るとレーンの一部が塞がり、サンプルをのせる時に苦労する。
II. 泳動サンプルの調製
- 暖めて均質にした4×loading bufferに1/20容の2-メルカプトエタノールを加える。[*13]
- 耐熱性のエッペンチューブ内で、loading buffer/蛋白質サンプル=1/3の体積比で混合する。[*14][*15]
- ビニルテープや市販のキャップロック(SSI社製他)でチューブのフタをしっかり留め、耐熱性のチューブフローターに差し込んで沸騰水に浮かべ、3分間加熱する。[*16]
- 「III. 電気泳動」へ進むか、すぐに泳動しない場合は−25℃で保存する。
【Tips】
- *13
- S-S結合の還元が不要な蛋白質はこの手順は不要。また、2-メルカプトエタノールを加えたloading bufferを−25℃でストックしても良い(数ヶ月は使用可能だが、凍結・解凍を繰り返さないこと)。
- *14
- 蛋白質濃度が薄いなどの事情でloading bufferの比率を下げたい時は、6×loading bufferを作る。要はFinalが10%グリセロール,2% SDS,0.1% BPB,0.05 M Tris-HCl, pH 6.8となれば良い。
- *15
- ★重要★レーンにのせるサンプルの体積や塩組成がなるべく揃うように調製すること。これらにバラつきがあると、バンドの歪みやにじみが起こり易い。
- *16
- 湯を沸かすのが面倒!サンプルチューブに水が触れるのは嫌!という現場の声が挙がり、筆者の研究室ではもっぱらヒートブロックで加熱(99℃,3分間)している。
III. 泳動
- ゲルを泳動槽にセットする。泳動槽下部に予め泳動bufferを入れ、ゲルの下部に気泡が入らないよう注意すること。
- [図2a] の要領でレーンに泳動サンプルをのせる。[図2b] のようにサンプル名を書いたビニルテープを泳動プレートに貼ると、想像以上に(視覚的にも精神的にも)楽にサンプルをのせることができる。特にサンプル数が多い時は効果大である。泳動終了後、このテープはゲル染色トレーのラベルに使える [図2c]。
- 分子量を調べたい場合は市販の分子量マーカーもレーンにのせる。
- 室温にて泳動する。始めはゲル1枚当たり15 mAの定電流で泳動し、泳動先端(BPB)が分離ゲルに移行したら150 V定電圧に切替える。[【解説】参照]
- 泳動先端がゲルを通り抜けてしまう前に(目的により通り抜けてもOKだが)泳動を止め、速やかに「IV. バンドの確認」へ進む。[*17]
ゲルの剥がし方、トレーへの落とし方は [図3] 参照。
【Tips】
- *17
- 速やかに “バンドの固定” 操作を行うこと。通電を止めた時点で蛋白質の拡散が始まり、長時間放置するとバンドがぼやけたり消失したりする。
【解説】定電圧or定電流?
定電流
- ほぼ等速で泳動が進むので泳動時間を予測しやすく、泳動条件を揃えやすい。
- 泳動が進むほど電圧が上昇し、ゲルの発熱量が多くなる。熱はバンドのスマイリングや蛋白質の変性の原因となるので、恒温式または冷却機能を有する泳動槽が必要。
- 複数枚数のゲルを同時に泳動する場合、「○ mA×枚数」で設定電流を計算するが、ゲル組成に違いがある場合は、当然ゲルごとに流れる電流が違ってくる。
定電圧
- 定電圧なので、想定外のゲルの発熱は起こらない。
- 泳動開始時に電流が最大で、以降徐々に電流が低下するので、濃縮ゲルと分離ゲルで電圧の切替えが必要。また、泳動の後半速度がどんどん遅くなるので、ややイライラする。
- 泳動するゲルの枚数に関係なく設定電圧を一定にできるので、うっかり屋さんがやっても失敗が少ない。
筆者の方針
- SDS-PAGE:蛋白質の熱変性は気にしなくて良いが、スマイリングを避けるために室温にて恒温式泳動槽で行う。濃縮ゲルでは定電流で電流を遅めにコントロールし、分離ゲルでは可能な範囲の電圧を一気に流して泳動を終わらせる。
- Native-PAGE:発熱による蛋白質の変性と長すぎる泳動時間の両方に注意が必要なので、低温室(4℃)にて恒温式泳動槽で行い、濃縮・分離ゲルとも定電流で泳動速度をコントロールして行う。
■ Tricine-SDS-PAGE ■
低分子量領域の分離を良くするには、ゲル(アクリルアミド)濃度を上げればよい。しかし、ゲル濃度が上がる程ひび割れや気泡の混入が起こり易く、ゲルの作成が困難になる。また泳動時の発熱のコントロールも難しい。Tricine-SDS-PAGEでは、先に示したSDS-PAGEよりも低いゲル濃度で、低分子量の蛋白質(0.3~1 kDa)を分離することができる。
試薬
- 3 M Tris-HCl, pH 8.45 [*18]
- 48%アクリルアミド-1.5%ビスアクリルアミド:遮光冷蔵保存 [*18]
- 80%グリセロール
- 他、10% SDS,10% APS,TEMED,50% 2-プロパノールはSDSゲルと同じ
- 泳動buffer(陽極側):0.2 M Tris-HCl, pH 8.9
- 泳動buffer(陰極側):0.1 M Tris-0.1 M Tricine-0.1%SDS [*19]
【Tips】
- *18
- これらの試薬とゲル調製に使う純水は0.45 μmのフィルターで濾過する。
- *19
- Trisはシグマ社製T1503を使用。pHは調整しない。
- *20
- ★重要★バンドの確認は銀染色またはCBB-Rで! グリセロールを含むゲルは、CBB-G(市販のゲル染色キット等)では染まり難い。
実験手順
ゲルの作成手順は基本的にSDSゲルと同じ。ゲルが [図4] のように3段になるので、以下のフローチャートに示すようにステップが増える。ゲル溶液の組成は [表2] の通り。泳動bufferが陰極側と陽極側とで異なるので、ゲルを泳動槽にセットする際は両bufferが混ざらないよう工夫すること。泳動は定電圧で、恒温式泳動槽を使用して行う。泳動サンプルの調製法はSDS-PAGEと同じ。
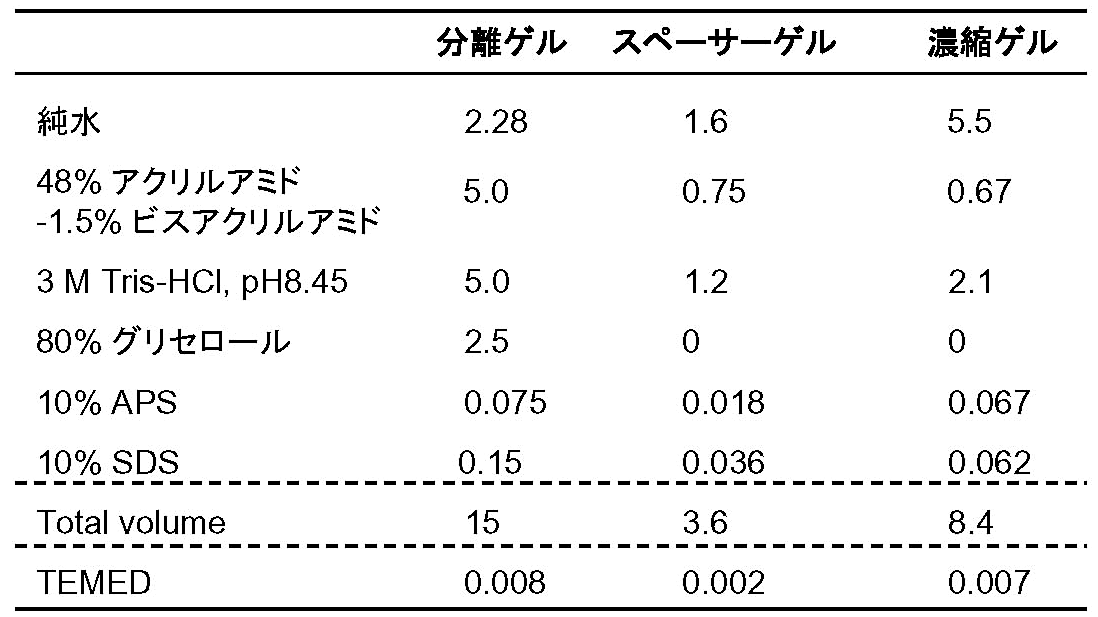
分離ゲルの重合(要50% 2-プロパノールの重層)[20分]
↓
泳動プレートの洗浄 [1分]
↓
スペーサーゲルの重合(要50% 2-プロパノールの重層)[20分]
↓
泳動プレートの洗浄 [1分]
↓
濃縮ゲルの重合(コウムを挿入)[25分]
↓
レーンの洗浄 [1分]
↓
蛋白質サンプルをゲルにのせる
↓
泳動(開始から30分間は50 V定電圧、以降は150 V定電圧)[ミニゲルで90分]
■ Native-PAGE ■ ミニ60分
蛋白質分子のゲル内移動速度に、三大ファクター「分子量」「形」「電荷」のすべてが反映される。よって、蛋白質の四次構造(単量体or多量体など)や分子コンパクトネス、電荷の状態など、条件を十分に検討すれば様々な情報が得られる。
試薬
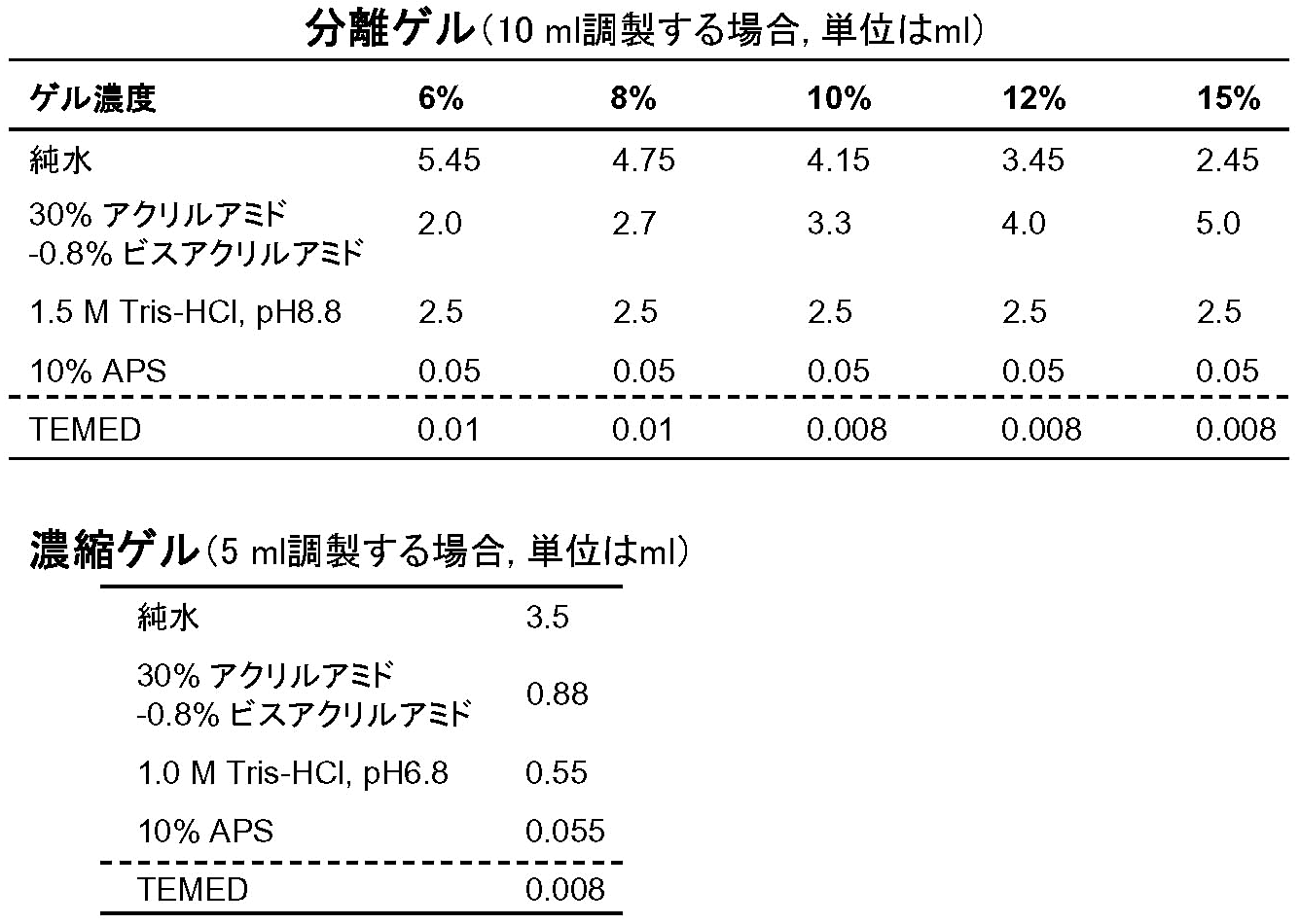
ゲル調製のための試薬:SDSゲルと同じ(SDS,2-メルカプトエタノールは不要)
- 泳動buffer(陽極側):0.1 M Tris-HCl, pH 7.8
- 泳動buffer(陰極側):0.068 Mグリシン-0.053 M Tris-HCl, pH 8.9
- 4×loading buffer:40%グリセロール-0.4% BPB-0.2 M Tris-HCl, pH 6.8 [*21]
【Tips】
- *21
- SDS用loading bufferのような使用時の加温は不要(むしろ厳禁)。泳動サンプルはloading buffer/蛋白質サンプル=1/3の体積比で混合して調製し、当然ながら加熱処理はしない。
実験手順
- ゲルの作成手順はSDSゲルと同じ。ゲル溶液の組成は [表3] の通り。
- ゲルは冷蔵保存可能だが、SDSゲルよりは短命。
- 泳動bufferが陰極側と陽極側で異なるので、ゲルを泳動槽にセットする際は両bufferが混ざらないよう工夫すること。
- 泳動は定電流で、低温室(4℃)にて恒温式泳動槽を使用して行う。
- 泳動先端(BPB)が濃縮ゲル中にある時はゲル1枚当たり10 mA、分離ゲルに移行したらゲル1枚当たり15~20 mA(電圧が180 Vを超えない範囲)の電流を流す。
- 低温室での実施が困難な場合は、冷蔵庫内で行うか、冷却水を還流できる泳動槽を利用してもよい。どうしても室温でしか行えない場合は、100 V定電圧で実施する。
■ 尿素-PAGE:濃度勾配なし ■
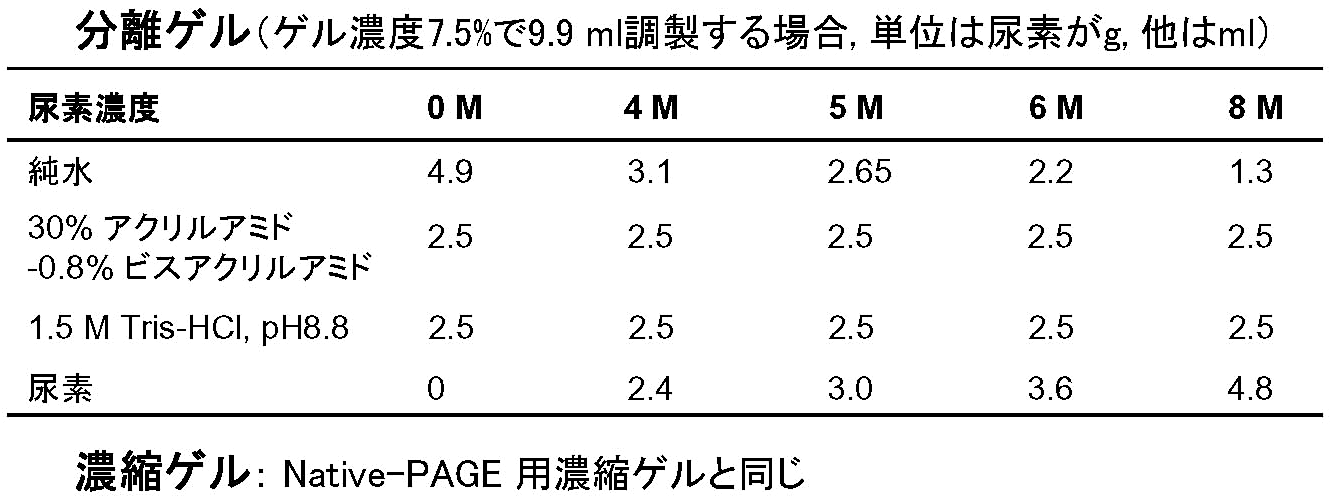
ゲル内の尿素が蛋白質の変性に十分な濃度の場合、「分子量」「形」「電荷」の三大ファクターの中で「形」が無視できるようになる。結果、同一分子量の蛋白質の電荷の違いを評価できる(リン酸化など)。また、Cys残基を化学修飾することでS-S結合の数を調べることができる [図5]。更に、尿素濃度を適切に設定すれば、蛋白質の安定性の評価もできる [図6]。
試薬
ゲル調製のための試薬、泳動buffer(陽極・陰極)、loading bufferはすべてNative-PAGEと同じ。
実験手順
- [表4] に従い、分離ゲル液を調製する。尿素は37℃くらいに温めて、ガラス棒などで緩やかに攪拌して溶かす。スターラーやボルテックスはなるべく使わない。
- 分離ゲル溶液の1/100容の10% APSを加え均一にした後、ゲル溶液の1/1000容のTEMEDを加えて泳動プレートに注ぐ。以下、2-プロパノールを重層するなど、他のゲルと同じ手順で分離ゲルを作成する。
- 濃縮ゲルの組成および作成手順はNative-PAGE用とまったく同じ。また、泳動bufferや泳動条件もNative-PAGEと同様である。ただし、分離ゲルに高濃度の尿素を含むので、ゲルは24時間以内に使用し、泳動中以外は低温に置かない。
■ 尿素-PAGE:尿素の濃度勾配あり ■
泳動方向に対いして垂直に(横軸方向に)尿素の濃度勾配をつけたゲル。わずかな蛋白質量で安定性の評価ができるので、尿素変性に関わる解析の予備実験として有用 [図7]。
装置・器具
- 円筒内径1 cmの密度勾配装置(サンプラテック社製04023など)
- 泳動プレートA,B [図8]
- 自家製スペーサーA,B,C [図8][*22]
- パワフルなマグネッチクスターラー
- 長さ10 mm未満の攪拌子2個(VWR社製58948-353など)
- ディスポの5 mlスポイト
- ガラスまたはアクリル製のプレート(20 cm×20 cmくらいが使いやすい)
- 翼付き留置針(テルモSV-18CLKなど。チューブ内径は大きめが良い)
- 強力なクリップ [図9]:ゲル1枚当たり4個必要
【Tips】
- *22
- 1 mmのアクリル板とアクリルカッター(どちらも¥300程度)をホームセンターで購入し、必要数を自作する。
試薬
- コーキング液:レンジ加熱可能でスポイトが届く大きさの広口ねじ口瓶に2 gアガロース,25 ml 1.5 M Tris-HCl(pH 8.8),75 ml純水を入れ、フタを緩めた状態で電子レンジで加熱し、アガロースを溶かす。
その他の試薬は「尿素-PAGE:濃度勾配なし」と同じ。
泳動サンプルの調製
Native-PAGEと同様、4×loading buffer/蛋白質サンプル=1/3の体積比で混合して調製する。1枚のゲルにのせる量は、蛋白質量30~50 μg(CBB染色)、体積150 μlくらいが適当である。
実験手順の詳細
以下に示す試薬の量は、筆者使用の泳動プレート [図8] に適用されるものである。
- 1.コーキング液のアガロースを電子レンジで溶かす。
- 2.泳動プレートを [図9ab] のように組み立てる。
- 3.コーキング液が60℃くらいに冷めたら、[図9] の手順でプレートをコーキングする。
- 4.アガロースが固まったら、プレートと器具を [図10a] のようにセットする。
- 5.[表4] に従い、0 Mおよび8 M尿素の分離ゲル液を調製する。
- 6.密度勾配装置のコックが閉じられているのを確認してから、3.1 mlの8 Mおよび0 M尿素ゲル液を、[図10b] に示した筒にそれぞれ入れる(逆に入れてはいけない!)。
- 7.両筒に10% APSを31 μlずつ入れ、攪拌子を回転させる。
- 8.この時点で、8 M尿素ゲル液が等圧を保つために少し留置針のチューブの方へ移行していればOK。もし流れていない場合は留置針の先を下げるか、[図10c] のように筒の口を押して空気を追い出し、ゲル液がスムーズにチューブへ移行できるようにする。
- 9.チップを付け、目盛りを0.5 mlにセットしたP1000のピペットマンを2本準備する。この後10~15の操作は全速力で行い、なるべく2人で作業する。以下、2人の分担をA,Bで示し、同時に行う操作は同じ番号で表記する。
- 10AB.両筒にTEMEDを3.1 μlずつ入れる(2人で同時に)。
- 11A.8 M側の筒の上口からピペットマンでゲルを0.5 ml抜き取り、泳動プレートに注ぐ。
- 11B.0 M側の筒の上口からピペットマンでゲルを0.5 ml抜き取り、チップから液が流出しないよう、安定な場所に置く。
- 12A.密度勾配装置のコックを開け、[図10c] のように筒の口を押し、2本の筒をつなぐ流路に溜まった空気を追い出す。
- 13B.0 M尿素ゲル液が8 M側の筒へ流れと同時に、留置針を泳動プレートの隙間に刺し込んでゲル液を流す。
- 14A.ゲル液の残量が少なくなって来たら [図10c] のように筒の口を押して、なるべくゲル液を出し切る。
- 15B.留置針をプレートから抜き、直ぐに流水中で密度勾配装置と留置針を洗って、器具内でゲルが固まるのを防ぐ。留置針のチューブ内は特に直ぐ固まるので、注射器などを使って水を押し流してやる。
- 15A.13Bで取り置きした0 M尿素ゲル液を重層する。次に2-プロパノールを重層する。
- 16.30分間室温で放置してゲルを重合させる。
- 17.クリップを外し、プレートからスペーサーAを抜き取る。
- 18.スペーサーを使ってアガロースを除き、洗瓶に入れた純水でゲルを洗浄する。
- 19.スペーサーBを [図11a] のように差込み、[図11b] のようにクリップでプレートを固定して立てる。
- 20.Native-PAGE用濃縮ゲル液を4 ml調製し、TEMEDを4 μl加える。これを分離ゲルの上へ注ぎ、直ぐにスペーサーCを差し込む。
- 21.30分間室温で放置してゲルを重合させる。
- 22.スペーサーBはそのままでスペーサーCだけを抜き、洗瓶に入れた純水でゲルを洗浄する。このゲルは24時間以内に使用し、泳動中以外は低温に置かない。
- 23.スペーサーBを付けたままプレートを泳動槽にセットし、[図11c] に示した部分に泳動サンプルをのせ、Native-PAGEと同様にして泳動する。
■ バンドの確認法(固定・染色・脱色)■
CBB染色、銀染色、市販キットによる染色について解説する。抗体による発色は本WEB内の「ウエスタンブロッティング」を参照。
- 経済性:CBB染色>銀染色>>市販キット>抗体発色
- 感度:抗体発色>銀染色>市販キット>CBB染色
- 結果確認の迅速さ:市販キット>銀染色>CBB染色>抗体発色
- 操作の手軽さ:市販キット>CBB染色>銀染色>抗体発色
- 筆者お勧め度:市販キット>>CBB染色;銀染色、抗体発色は目的に応じ実施
I. CBB染色(CBB-R)
試薬
- CBB(Coomassie Brilliant Blue R-250)
- CBB液(2.5 mg/ml CBB-45%メタノール,10%酢酸):500 mlもあれば10年は大丈夫
- 固定液(50%メタノール-5%酢酸)
- 脱色液(5%メタノール-7.5%酢酸):3 Lガロン瓶に作り置きしておく
手順
- トレーに100 ml強の固定液を入れ、[図3] のようにゲルを落とし込む。[*23] [*24]
- トレーをシェーカー等に置き、室温で30分間振とうさせる(速すぎるとゲルが割れるので注意)。
- 固定液を捨て、CBB液をゲルが浸る程度入れて、室温にてシェーカー上で振とうさせる。振とう時間の目安は、1 mmのミニスラブゲルの場合40分、ラージゲルや2 mmのゲルはその2倍くらいである。
- 先に柔らかいチューブを付けた駒込ピペットでCBB液を回収するこのCBB液は再利用可能なので捨てないこと。
- トレーに脱色液をたっぷり入れ、キムワイプを1~2枚入れて [*25] 再びシェーカーにて振とうする。キムワイプは時々新しいものと交換すること。1時間もすればバンドは確認できるが、きれいな結果を得るには脱色を半日くらい続けた方が良い。[*26]
【Tips】
- *23
- 試薬節約のため、トレーのサイズは底面積がゲルの2倍以内が目安。ここでは100 mlの試薬でゲルが十分浸る大きさのトレーを想定している。100 mlの液量でゲルが十分浸らない場合は固定液を増やせば良い。ただし、このステップはゲルの洗浄も兼ねているので、液量は100 mlより少なくしないこと。
- *24
- 手順1、2のステップを省略し、ゲルを直ぐCBB液に浸して固定・染色を同時に行うこともできる。しかし、この方法ではCBB液が劣化して繰り返し利用できない上、CB液の処分も面倒である。筆者は固定液による洗浄・固定を推奨する。
- *25
- キムワイプがCBBを吸着し、脱色液をきれいにする。
- *26
- 電子レンジを使用すると速く脱色できるが、敢えてここでは触れない。加熱しなければ脱色液も再利用可能。筆者は目の粗い濾紙で濾過した後ガロン瓶に入れて保存している。
II. 銀染色
感度、コストパフォーマスンともに優れ、1時間余りで結果が確認できるところが利点。一方、CBB染色よりも操作が煩雑で、銀イオンを含む廃液が出るため「お手軽」とは言えない。また感度が良すぎて余計なバンドが見えることもある。更に、CBB染色に比べ定量性が乏しく、デンシトメーターを使用した解析には不向き。
試薬(ミニゲル1枚染色時の必要量)
- 固定液(30%エタノール-10%酢酸,100 ml)
- 30%エタノール
- 増感液(37 mg \(\ce{Na2S2O3 \cdot 5H2O}\)+100 ml純水)[*26]
- 銀染色液(0.2 g \(\ce{AgNO3}\) +100 ml純水+0.075 ml 37% \(\ce{HCHO}\))[*27]
- 現像液(6 g \(\ce{Na2CO3}\)+0.5 mg \(\ce{Na2S2O3 \cdot 5H2O}\)+100 ml純水+0.045 ml 37% \(\ce{HCHO}\))[*27]
- 酢酸
【Tips】
- *27
- いずれも使用直前に調製。以下の「手順」の項で調製のタイミングを示す。
手順
- 固定液が入ったトレーにゲルを落とし込む [図3]。
- 30~60分間シェーカー上で振とうし、蛋白質を固定する。
- トレーの液を捨て、30%エタノール(50~100 ml)に入れ替えて10~15分浸透する。
- 上記3の30%エタノールによるゲルの洗浄操作を合計3回繰り返す。最後の振とうの間に増感液と銀染色液を調製する。
- 30%エタノールを捨て、増感液を注ぎ、手で1分間振とうする。
- 増感液を捨て、直ぐに純水を100 ml以上入れて、手で1分間振とうする。
- 上記6の純水によるゲルの洗浄操作を合計2回繰り返す。
- 純水を捨て、銀染色液を注ぎ、シェーカー上で15~30分間振とうする。この間に現像液を調製する。
- 銀染色液を捨て、純水を100 ml以上入れて、手で1分間振とうする。
- 純水を捨て、トレーに現像液を入れて、手で振とうする。2~15分くらいでバンドが現れるので、適当な強度になったら5~10 mlの酢酸をトレーに入れて反応を止める。酢酸を入れると二酸化炭素の泡がシュワッとでるので驚かないこと。
【Tips】
- 銀染色したゲルをゲルドライヤーで乾燥させるのはNG。真っ黒になる。
- 塩基性蛋白質は銀染色では染まり難い。発色しない場合は、ゲルを純水で洗浄後、CBB-G染色(市販キット等)やCBB-R染色を試す。
III. 市販キットによるバンドの発色
高コスト(筆者の研究室ではナカライテスク社製CBB Stain One (Ready To Use)を使用、ミニゲル1枚当たり¥110)ではあるが、価格に見合うメリットがある。CBB-GタイプとCBB-Rタイプがあり、前者は後者に比べて感度が劣り、酸性蛋白質の発色が悪いが、迅速に結果を確認ができるという大きなメリットがある(Nativeゲルで15分、SDSゲルでも30分)。結果が直ぐに分かるという利点は想像以上に電気泳動に対する煩わしさを軽減し、実験計画も立てやすくなる。頻繁に電気泳動を行う研究室には是非とも導入をお勧めする。
【Tips】
手順は勿論、基本的には各社マニュアルに従うが、以下のコツを挙げる。
- 染色液の節約のため、トレーは泳動プレートがギリギリ入る大きさのものを使用する [図3c]。ミニゲルの場合10 mlもあれば十分染色できるので、使用者には10 mlを厳守するよう、専用の秤量器具(筆者の研究室では15 mlファルコンチューブを使用)をキットとセットにして置いておく。目分量でトレーに注ぐと大目に入ってしまうので、とんでもなく高コストになる。
- SDSが染色を阻害するので、SDSゲルではこれを洗い流す操作が加わる。この時、なるべくタップリの純水でゲルを振とうし、純水を交換する際もタップリの純水でゲルを洗うと速く染色できる。
- 多くのキットでは、脱色操作は不要か、または純水で数分程度である。ただし、ゲルを純水に浸けたまま放置するとゲルがふやけてバンドがにじむことがある。しばらく放置する場合はトレーの液を各社マニュアルにあるオプションの脱色液(30%メタノール-10%酢酸など)に置き換えること。
- CBB-Gタイプの場合(多くの市販キットはCBB-Gタイプ)、グリセロールを含むゲル(Tricine-SDSゲル等)は染まり難い。
文献
- Laemmli, U.K., Nature, 227, 680–685 (1970)
- Shägger, H. & von-Jagow, G., Anal. Biochem., 166, 368–379 (1987)
- Lomas, D.A., et al., J. Biol. Chem., 270, 5282–5288 (1995)
- Dafforn, T.R., et al., Methods, 32, 150–158 (2004)
- Takahashi, N. & Hirose, M., Anal. Biochem., 188, 359–365 (1990)
- Takahashi, N. & Hirose, M., J. Biol. Chem., 267, 11565–11572 (1992)
- Mahadeva, R., et al., J. Biol. Chem., 277, 6771–6774 (2002)
- Gooptu, B., et al., Proc. Natl. Acad. Sci. U.S.A., 97, 67–72 (2000)
- Onda, M, et al., J. Biol. Chem., 280, 13735–13741 (2005)
改訂履歴
2024年3月24日 改訂
- 著者所属の情報の更新
- 細かい文言の修正
- P.7 Tips No.20に注意追加。
- P.13 Tipsに注意追加。
- P.14 染色に関する文章追加。
- P.14 Tipsに注意追加。
-
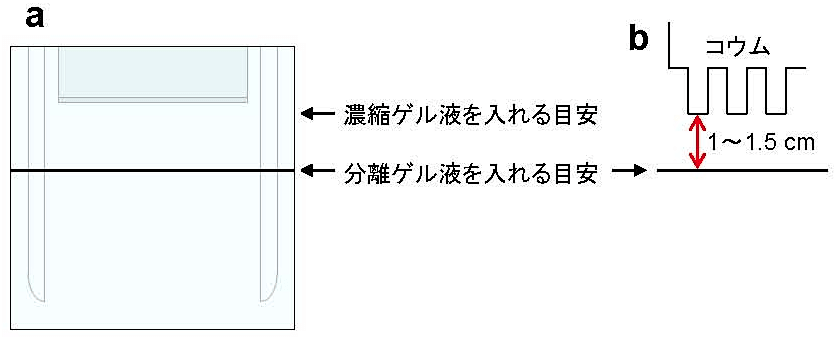
図1:泳動プレートの準備
分離ゲル液を入れる目安となる線を油性ペンで引く。ゲルや泳動bufferに直接触れない面に線を引くこと(a)。濃縮ゲルは1〜1.5 cm必要なので、プレートにコウムを差して線の位置を決める(b)。 -
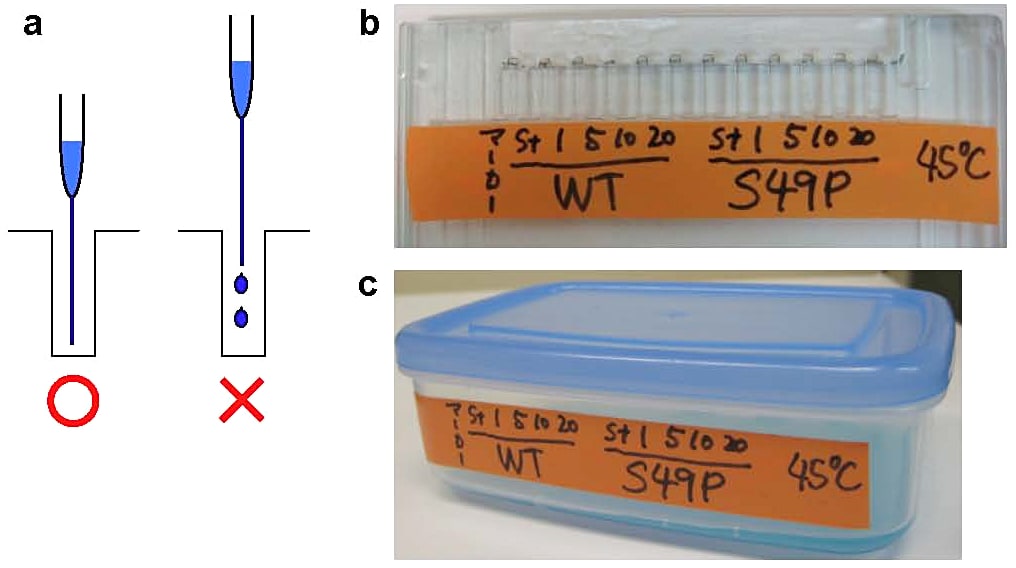
図2:泳動サンプルをゲルにのせる時のポイント
a. ローディングチップ(またはシリンジ)の先をレーンになるべく深く入れる(左)。浅いと泳動サンプルが拡散しやすい(右)。
b. サンプルの内容を書いたビニルテープをレーンの底が見えるギリギリのところに貼ると泳動サンプルをのせやすい。泳動終了後、このテープは染色トレーのラベルに利用できる(c)。 -

図3:泳動プレートからゲルを外す手順
a. 柄の先が平らなスパテラをプレートの角に差込み、片側のプレートを外す。
b. ゲルの両サイドに切れ目を入れる。濃縮ゲルを除く場合は濃縮ゲルと分離ゲルの界面に切れ目を入れ、スパテラで濃縮ゲルを上方へこそぎ落とす。
c. 固定液等を入れたトレーにプレートを斜めに入れ、スパテラでゲルの上底を少しだけ剥がす。するとゲルは重みで自然にトレーに落ちる。 -
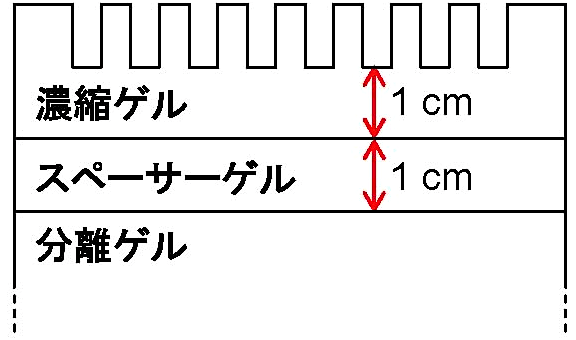
図4:Tricine-SDS-PAGE用ゲルの構成 -
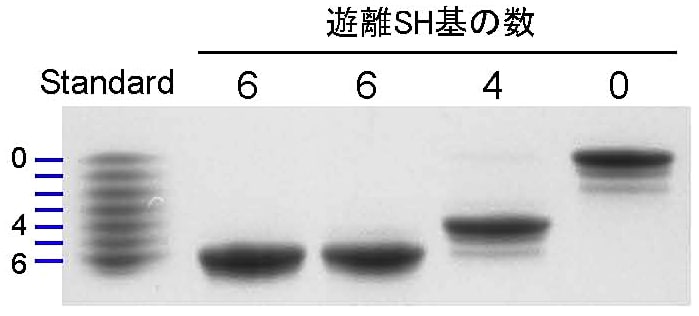
図5:尿素-PAGEによる解析例1:SH/S-S混合蛋白質の解析
まず、遊離のSH基をIAA(iodoacetic acid)でブロックする。その後、S-S結合をDTT(ditiothreitol)で還元し、これをIAM(iodoacetamide)でブロックする。得られたサンプルを8 M尿素を含むゲルで泳動すると、同一蛋白質であればS-S結合の数が多いほど泳動速度が遅くなる。(この手法の利用例:参考文献5,6)。
資料提供:高橋 延行 博士(京都大学農学研究科) -
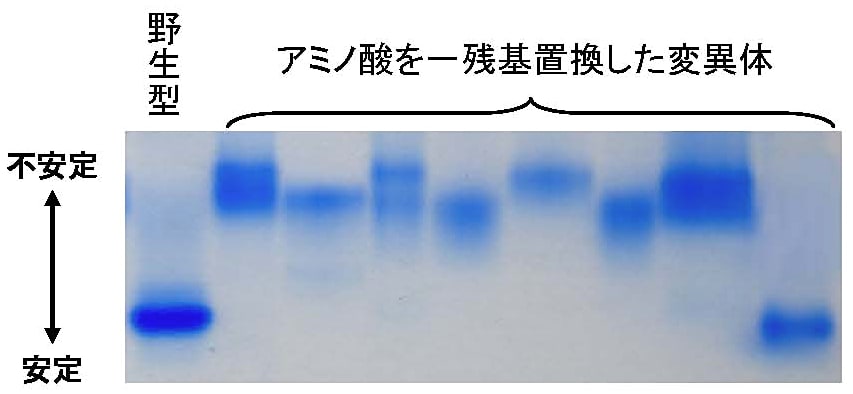
図6:尿素-PAGEによる解析例2
変異の導入部位により、尿素(3 M)に対して異なる安定性を示した。(この手法の利用例:参考文献7) -

図7:尿素濃度勾配-PAGEによる解析例
変異の導入により、2つの異なる安定性を示す分子種にフォールドした。
(この手法の利用例:参考文献8,9) -
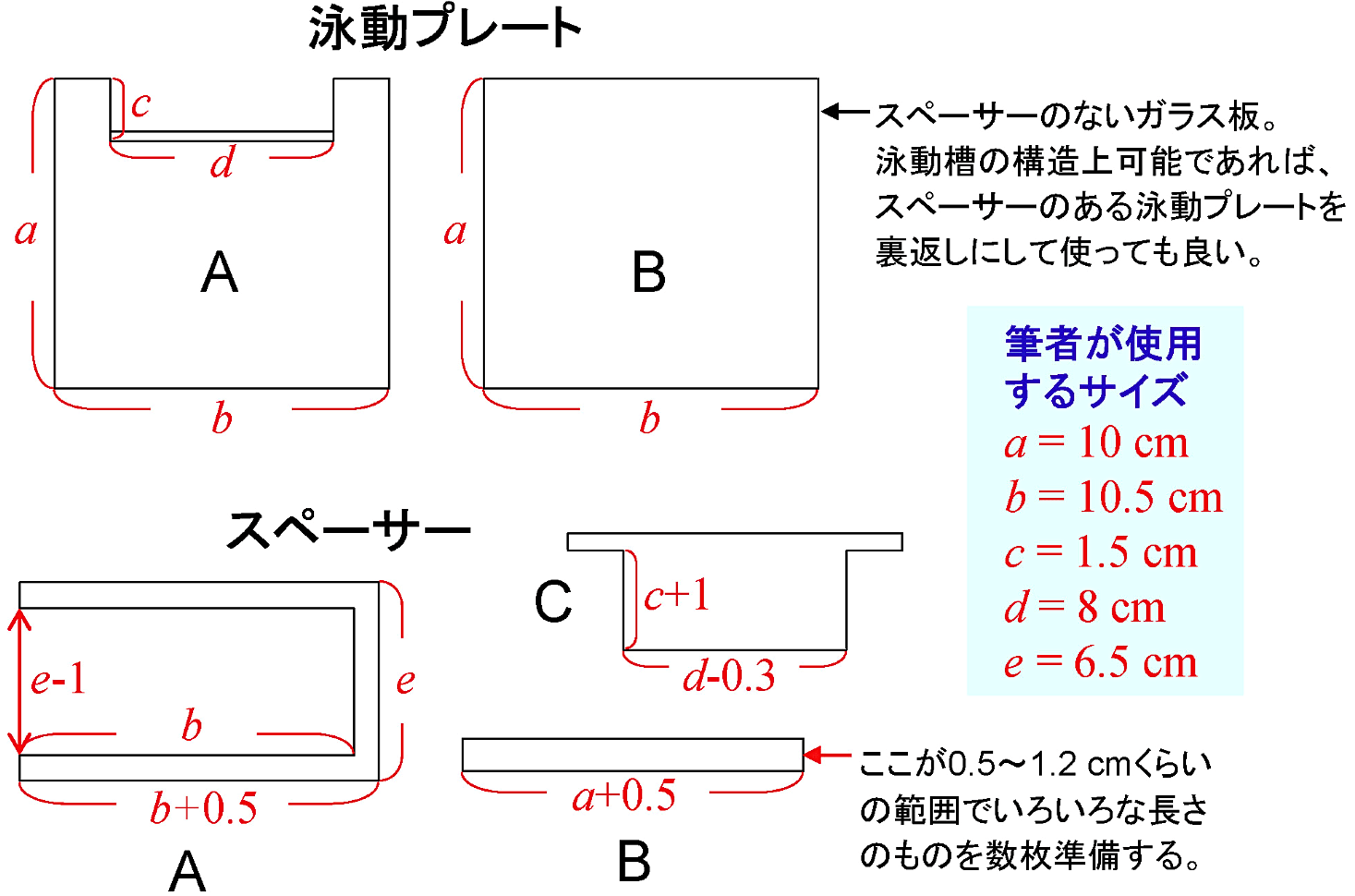
図8:尿素濃度勾配ゲル作成のためのプレートとスペーサー -

図9:尿素濃度勾配-PAGE:分離ゲル作成用プレートの準備
① 泳動プレートA,BとスペーサーAをaのように組立て、bのようにクリップで固定する。
② スポイトを使ってx,y,zの順にコーキングする(c)。
③ zの辺を下にしてガラスプレートに立て、底辺をアガロースで十分にコーキングする。 -

図10:尿素濃度勾配-PAGE:分離ゲル作成のための器具の配置
a. 密度勾配装置の2つの筒に1つずつ攪拌子を入れ、留置針を付ける。密度勾配装置は、留置針の先がギリギリ泳動プレートに刺し込める高さまで上げる。
b. ゲルを泳動プレートに注ぐまでは留置針の先は筒口より高い所に固定しておく。8 M尿素ゲル液は手前、0 M尿素ゲル液は奥の筒に入れる。逆は不可。
c. 親指で押して空気を追い出す操作。 -
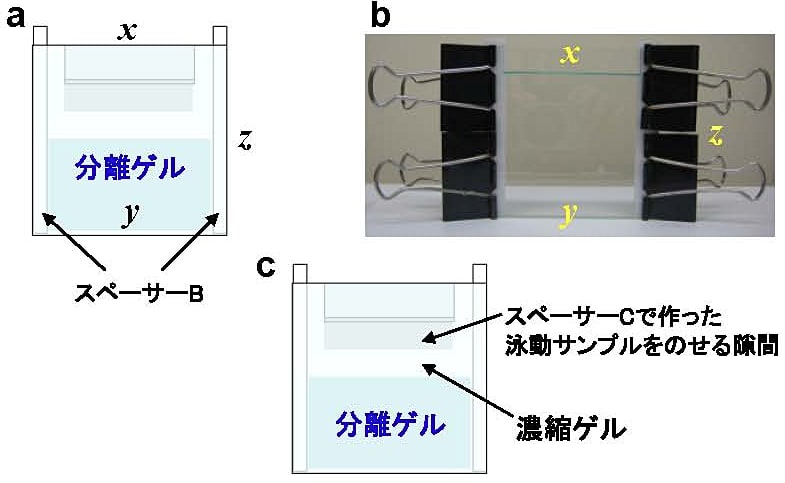
図11:尿素濃度勾配PAGE:濃縮ゲルの作成
a. 分離ゲル重合後、スペーサーBを差し込む。
b. プレートをクリップで固定して立てる。
c. 完成後のゲルのイメージ。
