

概要
近年、個々の生物種のゲノム配列が次々と決定されている。興味深いことに全遺伝子に対する膜タンパク質の数の占める割合は大腸菌からヒトに至るまで、おおむね30%にも及ぶ。しかしながら、膜タンパク質は水にほとんど溶けないリン脂質膜に埋もれているため、その物理、化学的性質は水溶性タンパク質と大きく異なり、そのことが研究上の困難となって、水溶性タンパク質ほど研究が進んではいない。個々の膜タンパク質を「タンパク質科学」するためにはまず、膜タンパク質、リン脂質を界面活性剤で可溶化し、分離、精製しなければならない。また、膜タンパク質の機能解析を行う際には、リン脂質二重膜(リポソーム)に再度埋め込むことが必要な場合もある。
本プロトコールではジフテリア菌のヘムセンサーキナーゼChrS(1)にヒスチジンタグ(His-tag)を融合した組換え膜タンパク質を大腸菌で発現させ、可溶化、精製、およびリポソームへの再構成を行うための手順を紹介したい。
装置・器具・試薬
- 大腸菌BL-21(DE3)pLysS
- ベクターpRSET-A
- 遠心機(各社)
- 超遠心機(各社)
- フレンチプレス(大岳製作所)
- Ni-NTA 樹脂(QIAGEN社)10 mL
- 界面活性剤
- OG:オクチルグルコシド(n-octyl‐β‐glucoside)
- SM:シュクロースモノラウレート(sucrose monolaurate)
- DDM:ドデシルマルトシド(n‐dodecyl‐β‐D‐maltoside)
- DM:デシルマルトシド(n‐decyl‐β‐D‐maltoside)
- プレシジョンプロテアーゼ(GE Healthcare社)
- 大腸菌リン脂質(Avanti Polar Lipids社)
実験手順
- 細胞膜の調製(0.5日)
- 可溶化、精製(3–4日)
- リポソームへの再構成(2日)(リポソームの調製は予めしておく、2日)
実験の詳細
膜蛋白質の調製
細胞膜の調製
第1日
-80℃で保存した菌体10gに対して200 mLの菌体破砕バッファー(30 mM Tris HCl pH 8、20%(w/v)sucrose)を加えて、ペレットを完全に分散させる。ここに1 mLの20 mg/mL lysozyme、2 mLの0.5 M EDTA-Naを添加し、4℃で15分間攪拌する。続いて15,000 rpm(34,000 g)で15分間遠心し、上清を50 mL捨てる。バッファーと沈殿した菌体を取り出し、1,200kg/cm2で2回French Pressを行う。破砕した菌体溶液と等量の3 mM EDTAを加えた後に1/300量の300 mM PMSF(phenylmethylsulfonyl fluoride;52 mg/mL DMSO)を添加する。12,000 rpm(22,000 g)で15分間遠心する。上清を回収して超遠心用のチューブに移し、ベックマン45Tiローターを用いて40,000 rpm(200,000 g)、4℃で1時間超遠心する。超遠心の上清を捨て、ガラス棒でペレットを回収し、タンパク濃度がおよそ20–30 mg/mLになるように細胞膜保存バッファー(25 mM Tris HCl pH 7.5、10%(w/v)sucrose)を加える。そして氷冷しながらホモジナイズし、溶液を- 80℃で保存する。菌体10 gから約200 mgの全膜タンパク質が得られる。
可溶化、精製
その前に
なんといっても、対象となる組み換え膜タンパク質が大腸菌細胞膜で機能をもった状態で発現されていることが望ましい。本研究ではChrSがヘム依存的なリン酸化活性を保持していることを確認済みである。次に、可溶化の条件検討が必要である。マイルドな非イオン性界面活性剤4種類OG、DM、DDM、SMで可溶化を試みた。膜タンパク質画分の終濃度が2 mg/mL となるように 2×可溶化バッファー(50 mM Tris-HCl pH 7.5、10% glycerol、300 mM NaCl)、Milli-Q水、界面活性剤(終濃度が1.2%)を加えた。4℃で1時間攪拌した溶液を200,000 g、4℃で1時間超遠心し、上清を回収した。そして可溶化されたタンパク質を SDS-12.5% PAGE によって確認した。図1よりオクチルグルコシドではChrSタンパク質は可溶化できないことがわかる。本研究では、再構成の際に透析で取り除きやすいDM(臨界ミセル濃度0.08%)を用いることにした。
第1日
200 mgの全膜タンパク質を1.2% DMで可溶化して得た上清100 mLをバッファーA(25 mM Tris-HCl pH 7.5、5% glycerol、150 mM NaCl、0.2% DM)で平衡化したNi-NTAカラム(bed volume 10 mL)に添加する。バッファーの流速が1 mL/minとなるように調節し、100 mLのバッファーAでWashする。続いてカラムにバッファーA 80 mLとバッファーB(バッファーAにイミダゾール400 mMを含む)80 mLを、グラジエントをかけながら添加してChrS-PreScission-10xHisを溶出する(図2)。
第2日
得られたフラクションに関してSDS-PAGEを行い、ChrS-PreScission-10xHisに相当するバンドが確認されたフラクションのみ回収する(10–15 mg)(図3;レーン2)。濃縮器セントリプレップYM-50(Millipore社)を用いて、タンパク濃度が1–2 mg/mL程度になるまで2,500 rpm,4℃で濃縮し、ChrSの1/100重量のPreScission protease(1unit/μg)を加える。この溶液を透析チューブに入れ、1,000 mlの透析バッファー(50 mM Tris-HCl pH 7.5、5% glycerol、150 mM NaCl、0.2% DM)、を用いて4℃で3時間透析を行い、3時間後に別の透析バッファー1,000 mLに移し、一晩透析をする。
第3日
Protease処理後の、透析チューブの中身の一部(2–4 mg相当)をSDS-PAGEにかけ、切断を確認した後(図3;レーン3)、その溶液をNi-NTA affinityカラム(bed volume 10 mL)に直接添加した。Flow throughとバッファーA 10 mLでのWash画分を回収し、さらにバッファーA 50 mLとバッファーB 50 mLでグラジエントをかけながら溶出された画分も回収した。そして各フラクションをSDS-PAGEに流して、His-tagが切断されたChrSが確認されたフラクションのみ回収し、タンパク濃度がおよそ2.5 mg/mLになるまでセントリプレップYM-30を用いて2,500 rpm(1,100 g)、4℃で濃縮する。最終的なタンパク濃度を測定して-80℃で保存する。濃縮が終わらなかったら翌日に続ける。Hig-tagを切断した最終精製標品を図3のレーン4に示す。
リポソームへの再構成
大腸菌リン脂質からのリポソーム調製
第1日
Avanti社大腸菌リン脂質はクロロホルムに溶解している。まず40 mg分のリン脂質を遠心エバポレーターで減圧濃縮し、クロロホルムを除去する。1.5%オクチルグルコシドを含む 50 mM Tris-HCl pH 7.5 溶液を5 mL加え、ボルテックスで溶解させる。溶けない場合はオクチルグルコシドを3%にする。次に、透析バッファー(50 mM Tris-HCl pH 7.5)を1,000 mL用いて3回透析し、界面活性剤を取り除くことでリポソームを調製する。
第2日
リポソーム試料液の容積を測り、リン脂質濃度を調べる(6–8 mg/mLとなる)。エッペンドルフチューブに分注して液体窒素で凍らせ、室温でゆっくり溶かすという作業を2回繰り返し、最後に再度液体窒素で凍らせて-80℃に保存する。リポソームを使用する時には室温でゆっくり溶かして用いる。
再構成
第1日
| ChrS (2.4 mg/mL) | 0.05 mL |
| 大腸菌リポソーム (8 mg/mL) | 0.3 mL |
| 再構成透析バッファー | 0.115 mL |
| 10% OG | 0.03 mL |
| 1M DTT(dithiothreitol) | 0.005 mL |
| total | 0.5 mL |
溶液を調製後15分間室温で放置し、透析チューブに移す。その後500 mLの再構成透析バッファー(50 mM Tris-HCl pH 7.5、50 mM KCl、5% glycerol)を2つ用意し、4℃で2回(1回目は3時間、2回目はオーバーナイト)透析する。
第2日
再構成したプロテオリポソームを回収し、分注して-80℃に保存する。
このようにしてリポソームに再構成したChrSはヘムに依存した高い自己リン酸化活性をもっていることがわかる(図4)。
工夫とコツ
発現系について
本プロトコールではベクターpRSET-Aの多コピー性、T7プロモーターのみを利用する。つまり、もともとベクターに付与されているHis-tagやプロテアーゼ切断部位は取り除く。代わりに、ChrS-プレシジョンプロテアーゼ切断配列-10×Hisタグを連結したDNAをNdeI、HindIII部位に連結する。
ChrSはN末に6回膜貫通領域をもち、C末が水溶性ヒスチジンキナーゼドメインである。そこで、細胞膜への局在を損ねないように、His-tagはC末に融合する。C末にタグを付加する場合には、プレシジョンプロテアーゼ切断配列を付加してあるものの、His-tag切断後も一部の配列Leu-Phe-Glnが残ってしまうのが欠点ではある。しかし、結果的にはChrSの機能に影響はないようである。
大腸菌膜タンパク質にはNi-NTA樹脂に吸着し、100 mM程度のイミダゾールで溶出されるタンパク質群がある(しかも、よりによってヘムタンパク質も含まれている)。そこで組換えタンパク質のNi-NTA樹脂への保持力を高めるために、通常の6×Hisではなく、10×His-tagを付加し、高濃度イミダゾールで溶出させることで分離を高めた(図2)。
培養について
前培養は本培養の1/20量のLB+0.5%グルコース培地で行う。本培養はTB培地で25℃、一晩で行う。イソプロピル-β-チオガラクトピラノシド(IPTG)は加えない。1 Lあたり、10–14 g湿重量の菌体が得られる。菌体は-80℃で保存する。
大腸菌にとって有害なタンパク質は前培養では発現させないほうがよい。そこで、T7 RNAポリメラーゼを発現させないように0.5%グルコースで培養している。また、筆者はグルコースがプラスミドコピー数も少なくしているような気がする。本培養ではIPTGを加えない。あまりにも大過剰な発現は正しくフォールドしたタンパク質を与えず、活性を失っていたり、可溶化できないことがある。
界面活性剤の除去方法
リポソームへの再構成とは界面活性剤ミセルに覆われた膜タンパク質を再びリン脂質二重膜に埋め戻す操作である(2)。この操作には界面活性剤を取り除く作業が含まれる。本研究では透析法を用いたが、オクチルグルコシドのような臨界ミセル濃度が極めて高いもの(0.7%)では希釈-遠心法も有効である。また、ドデシルマルトシドやトライトンX100などではプラスチックビーズ(BioBeads、バイオラッド)を加えて、界面活性剤を吸着させて取り除く方法もよく利用されている。
文献
- Schmitt, M.P., J Bacteriol, 181, 5330–40 (1999)
- Rigaud, J.-L. & Levy, D., Methods in Enzymology, 372, 65–86 (2003)
-
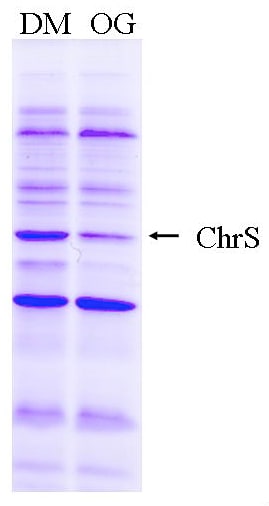
図1:デシマルトシド、オクチルグルコシドで可溶化した大腸菌膜タンパク質
ChrSタンパク質はオクチルグルコシドではほとんど可溶化できないことがわかる。 -
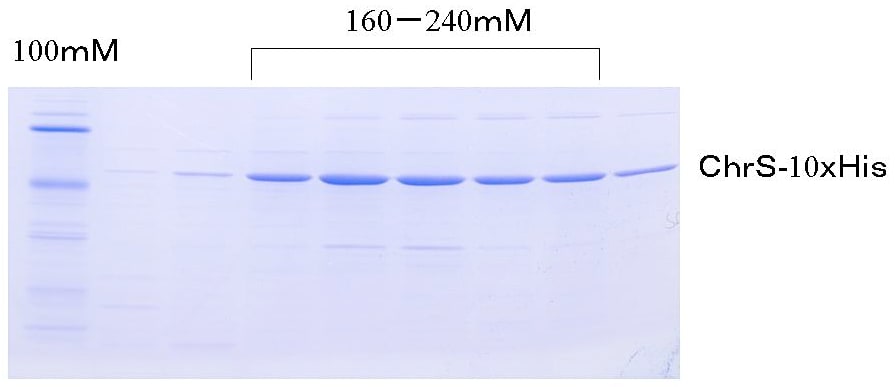
図2:デシマルトシドで可溶化した大腸菌膜タンパク質のNi-NTAカラムからのイミダゾールによる溶出
100 mM付近のイミダゾールで大腸菌由来の膜タンパク質が多数溶出される。6×ヒスタグタンパク質は重複して溶出されるが、10×ヒスタグタンパク質は遅れて溶出される。 -

図3:ChrSタンパク質の精製 -
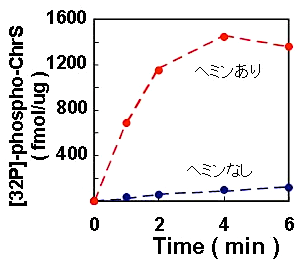
図4:リポソームに再構成したChrSタンパク質のヘム依存的自己リン酸化活性
