
研究室メンバーで一丸となってソフトウェアを開発するには?
理化学研究所・杉田理論分子科学研究室- 森貴治, 蛋白質科学会アーカイブ, 12, Essay 07 (2019)
- https://www.pssj.jp/archives/essay/Es_07/Es_07.html

この度は日本蛋白質科学会若手奨励賞・優秀賞を頂き大変光栄に思います。今回、クライオ電顕データと MD 計算に基づくタンパク質の立体構造モデリング法(フレキシブル・フィッティング)に対して高速並列計算法を考案し、リボソームなどの巨大生体分子複合体への応用を可能にしました (1)。開発した手法は MD 計算プログラム GENESIS (2) に導入され、チュートリアルやマニュアル等と合わせて Web サイトにて公開される予定です (3)。実験研究者にも広く利用して頂ければ幸いです。GENESIS の開発は2010年頃から始まり、現在では解析ツールも含めて25万行に及ぶ巨大なプログラムパッケージになりました。私自身、初期の段階から開発に携わり、GENESIS の進化をリアルタイムに見てきました。このエッセイでは、このような巨大プログラムを研究室メンバーとどのように作成しているのかについて、特に「共同開発の大変さ」に焦点を当てて、開発者の一人として話をさせて頂ければと存じます。
そもそも巷には色々な MD 計算プログラムが既に存在するのに、なぜ新しいものを一から作ったのかと疑問に思うかもしれませんが、それは単純で、自前でプログラムを作成しているとソースコードを隅々まで理解しているので、新しい方法論を開発しやすくなるからです。従来にない手法を考案し、自分たちの興味のある系、例えば膜タンパク質などに適用することで新しい生命現象の発見や理解につなげることを目的としています。研究室メンバーが一丸となって開発をする理由は、後発である GENESIS が「機能と速度」の両面において GROMACS (4) や NAMD (5) などの外国産プログラムに対抗して世界一になることを目指しているためです。実際に「京」コンピュータを用いた細胞環境の1億原子系のシミュレーションでは、大規模計算を得意とする NAMD を遥かに凌駕する性能を GENESIS は示しました (2)。機能においてもさらに充実させる予定で、クライオ電顕フィッティング以外にも QM/MM 計算や自由エネルギー解析ツールなどが順次追加されることになっています。
ところで、開発者の人数が多くなればなるほど、プログラムの管理が容易でないことは想像するに難くないと思います。例えば、複数の人がソースコードの同じ箇所を変更する場合や、自分の修正が他人の開発に影響を及ぼす場合は、メンバー同士で議論をして意思疎通を図りながら開発を進める必要があります。それでもプログラムは日々様々な箇所で修正を受けているため、全メンバーがそれらを全て把握することは到底できません。そこで私たちは GitHub (6) を使って GENESIS を管理しています(図1)。これは近年ソフトウェア開発のプラットフォームとして世界中で用いられているツールで、Web 上で容易にプログラムの開発状況を知ることができます。開発者は GitHub サーバからプログラムの最新版をダウンロードし、各自の計算機端末でソースコードを修正した後、サーバにアップロードすることでプログラムを更新していきます。複数の人が同じ場所を修正してアップロードしようとした場合、最初にアップロードされたものが優先され、それ以降のものには衝突判定がなされます。衝突が起こっている箇所を注意深く修正した後に再アップロードすることで最新版が更新されることになります。また、論文として出せるような方法論が各メンバーによって複数同時に開発され、それらをマージするような大規模な更新を行う際には、論文が出る順番も考慮しながら慎重に作業を進めています。
巨大なプログラムともなると、1カ所の修正が別の場所に予期せぬ影響を与えることもあります。例えば、A さんが新たに機能を追加したことによって、B さんの開発した機能に影響を与え、B さんの機能で誤った結果を出力してしまうようなケースです。それを簡便に検知するための策として、私たちはテスト計算用サーバを構築しています(図1)。GitHub 上でプログラムが更新されると、自動的にテストサーバがプログラムをダウンロードし、予め用意しておいたテスト計算を実行します。様々なパラメータを組み合わせた網羅的なテストセットを用意しており、更新前のプログラムで得られた結果と少しでも不一致が生じると、開発者全員にアラートメールが送信されます。不一致が起きたパラメータセットを調べることで自分の更新がどの機能に影響を与えてしまったかを確認できます。このような仕組みによって開発者は安心して GENESIS に新規機能を追加することができるのです。
1つのプログラムを共同して作成するには、各自のプログラミングスキルがある一定レベル以上であることも重要です。GENESIS は Fortran 言語で書かれており、また「京」コンピュータでの利用を目的として開発されたという経緯もあって、大多数の CPU を用いて効率良く計算するためにハイブリッド MPI/OpenMP 並列計算プロトコルが導入されています。MPI は主に CPU 間での並列計算(分散メモリ型並列)、OpenMP は CPU 内での並列計算(共有メモリ型並列)に利用され、マルチコア CPU 上で通信量を抑えた効率の良い演算を実現しています。さらに近年 GPGPU を用いた演算が科学技術分野で主流になり始め、このような状況に対応すべく GENESIS では計算コストのかかる van der Waals 力やクーロン力などの非共有結合性相互作用の計算を GPGPU で加速させています (7)。当然ながら GENESIS 開発陣には並列計算プログラムを「読める」だけでなく、「書ける」ことが要求されます。私自身、開発当初は並列計算プログラミングの経験が少ししかありませんでしたが、共同開発者で並列計算が得意な Jaewoon Jung 博士が書いたコードを見て勉強し、プログラミング講習会に参加するなどしてスキルを身に付けてきました。また Jung 氏も Fortran 言語は初めてだったらしく、GENESIS のコードを読みながら勉強したそうです。このように開発者は全員が始めから完璧なプログラミングスキルを持っているわけではないので、無いものは積極的に身に付ける努力も必要となってきます。
GENESIS 開発の理念の1つとして、ソースコードを誰が見ても理解できるように分かりやすく書き、誰でも簡単に新規機能を追加できるプラットフォームにすることを掲げています。そのための工夫として、GENESIS は2つの異なる MD 計算プログラム ATDYN と SPDYN、それからトラジェクトリー解析ツールを用意しています。ATDYN は原子分割法(Atomic decomposition)、SPDYN は空間分割法(Spatial decomposition)で並列化されており、前者は原子間相互作用ペアを CPU 数に応じて分割するのに対して、後者はシミュレーションのボックスを CPU 数に応じてドメインに分割します(図2)。原子分割法に比べて空間分割法では大規模計算における通信コストを抑えられるため、通常、ATDYN はペプチドや小さなタンパク質、SPDYN は膜タンパク質などの計算に用います。原子分割法では全 CPU が系に存在する全ての原子の座標情報を保持しているため、比較的容易にコードを理解でき、開発初心者にとっては CPU 間の通信法をあまり気にせずに新規機能を追加することができます。一方、空間分割法では各 CPU が割り当てられた領域内の原子座標の情報しか保持しておらず、「隣り合うドメイン間で座標を送信して力を受信する」という通信を行うため、コードに詳しくなければどのように原子間相互作用を計算すればよいか分からないという事態に陥ります。しかし、ATDYN と SPDYN を両方用意することで、開発に新たに加わったメンバーは、まずは ATDYN をしっかりと理解することで MD 計算の全体像を把握する修行に勤しみ、さらに難しい SPDYN へとスムーズに移行できる仕組みになっています。Fortran 言語に慣れていない人は、場合によっては ATDYN よりもさらに簡単な解析ツールから出発することになります。杉田有治主任研究員によると、これをサッカーの名門チーム FC バルセロナに例えて、ATDYN を改変できる人はバルサ B チーム(FC バルセロナ B)、SPDYN まで改変できれば A チームに昇格できるというわけです。実際チームに分かれているわけではありませんが、プログラムへの理解を深めることで徐々に昇格でき、より困難な研究に挑戦することが可能になるのです。
ちなみに GENESIS のロゴは私が作成したもので、箱が格子状に分割されているのはこの空間分割法をイメージしています。ロゴをすべて直線で描き、GENESIS の最初の3文字 G, E, N を箱表面にタイル状に貼ることでソフトウェアとしてのデジタル感を出しています。GENESIS の I だけ色が違うのはちょっとしたオシャレのためであって特に意味はないのですが、敢えて I を選んだ理由はその下に水平線を入れたのでバランスを取るためです。当初は京コンピュータのイメージカラーと同じ朱色を用いようと思ったのですが、意外と難しくて似たような色にしたというのが経緯です。
一つ開発初期の段階の苦労話をしますと、私が最も時間を費やした作業の一つは、CHARMM や NAMD などの既存ソフトウェアとエネルギーや力の値を比較して、倍精度(有効数字15桁程度)で完全に一致する結果を出すことでした。数万行あるプログラムの中で単純に計算の符号を間違えたり、温度や圧力制御の際に速度と座標の更新の順番を間違えたりすると、シミュレーション中に系が爆発することもあり、ありとあらゆる方程式を正しく解かなければならないことを実感します。爆発というのはシミュレーション中にボックスサイズが急激に大きくなり、溶媒分子が蒸発して飛散するような状況です。MD 計算プログラムをゼロから開発したことがあるならば、ほとんどの人が経験することだと思います。かつて NPT アンサンブル下において CHARMM 力場を用いてテスト計算を実行した際、脂質二重膜は正しく計算できるのに、膜タンパク質を二重膜に埋め込むとなぜか系が爆発するという奇妙な問題に直面したことがあります。その原因はどこにあったかというと、CHARMM 力場の CMAP の計算、つまりタンパク質主鎖の二面角エネルギーの補正項由来のビリアルの符号だけが逆になっていて、正しく圧力が計算できていなかったことに起因していました。非常に単純な間違いですが、確かに合理的な結果を出力したことになります。GENESIS はこのように数多くの細かい修正を経て現在の安定版に至っているのです。
最後に、GENESIS が今後どのような方向に進んでいくかについて話をすると、いくつかあるうちの1つはやはりタンパク質複合体の精密な立体構造モデリングへ向けて機能を拡張していくことが挙げられます。近年、クライオ電顕を用いることでタンパク質の近原子解像度の立体像が実験で得られるようになり、技術革新によりさらに解像度が向上してきましたが、タンパク質中の揺らぎの大きいドメインは一般的に局所解像度が低くなるため、このような部位に対しては実験データに大きく頼らずに計算科学の力を借りて構造を予測する必要も出てきます。ただし、粗い情報や平均化によって縮退した情報から詳細な情報を抽出することは「逆問題」を解くことになり、そのような実験データから唯一の構造を求めることは理論的に不可能です。その為には実験データをアンサンブル平均として理解する、あるいはノイズや誤差の影響も考慮する必要があり、ベイス推定などを用いて確率的に実験データと一致する立体構造を求める方法論も開発されています (8)。さらにクライオ電顕と他の実験データ、例えば、クロスリンク質量分析や SAXS, FRET, 高速 AFM データなどの低次元構造情報を、計算科学を駆使して組み合わせ、巨大な生体分子複合体構造を精密に組み立てることを目的とする新しい構造生物学 “Integrative Structural Biology” も提唱されています (9)。今後、計算科学による複合体の立体構造モデリングは益々重要となり、GENESIS が構造生物学の発展に少しでも貢献できればと考えています。
謝辞
GENESIS は理研・杉田有治主任研究員のもと、Jaewoon Jung 博士、小林千草博士、松永康佑博士、八木清博士、尾嶋拓博士らの多くの研究室メンバーと共同して開発しているプログラムであり、また、クライオ電顕フィッティング計算に関しては、理研・宮下治博士、Florence Tama 博士、Marta Kulik 博士と共同で行われたものです。今回受賞に至った成果は多くの方々によるプログラム開発が結集したことによりもたらされたものであり、この場をお借りして改めて御礼申し上げます。
文献
- Mori, T. et al., Structure, 27, 161–174 (2017)
- Jung, J.*, Mori, T.* et al., WIREs Comput. Mol. Sci., 5, 310–323 (2015) (*Equally contributed)
- https://www.r-ccs.riken.jp/labs/cbrt/
- Hess, B. et al., J. Chem. Theory Comput., 4, 435–447 (2008).
- Phillips, J. C. et al., J. Comput. Chem., 26, 1781–802 (2005).
- https://github.co.jp/
- Jung, J. A. et al., J. Chem. Theory. Comput., 12, 4947–4958 (2016).
- Bonomi, M. et al., Curr. Opin. Struct. Biol., 42, 106–116 (2017).
- Ward, A. B. et al., Science, 339, 913–915 (2013).
-
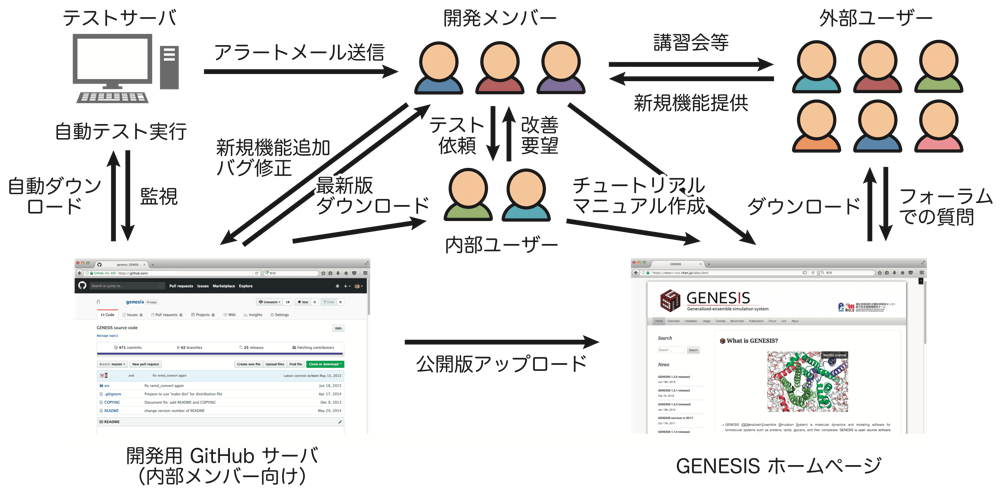
図1: GENESIS 開発実施体制。研究室メンバーは普段 GitHub 上の最新版プログラムを利用して研究を行っている。ホームページに公開されているプログラムは、開発版よりもおおよそ1〜2年程度遅れたものであるが、バグ修正と新規機能に対するテストが十分になされた安定版である。外部ユーザーはホームページのフォーラムに日本語でも投稿でき、開発陣が疑問などに答えてくれる。また、外部ユーザーとも連携して新規機能を導入できるような体制を取っている。 -
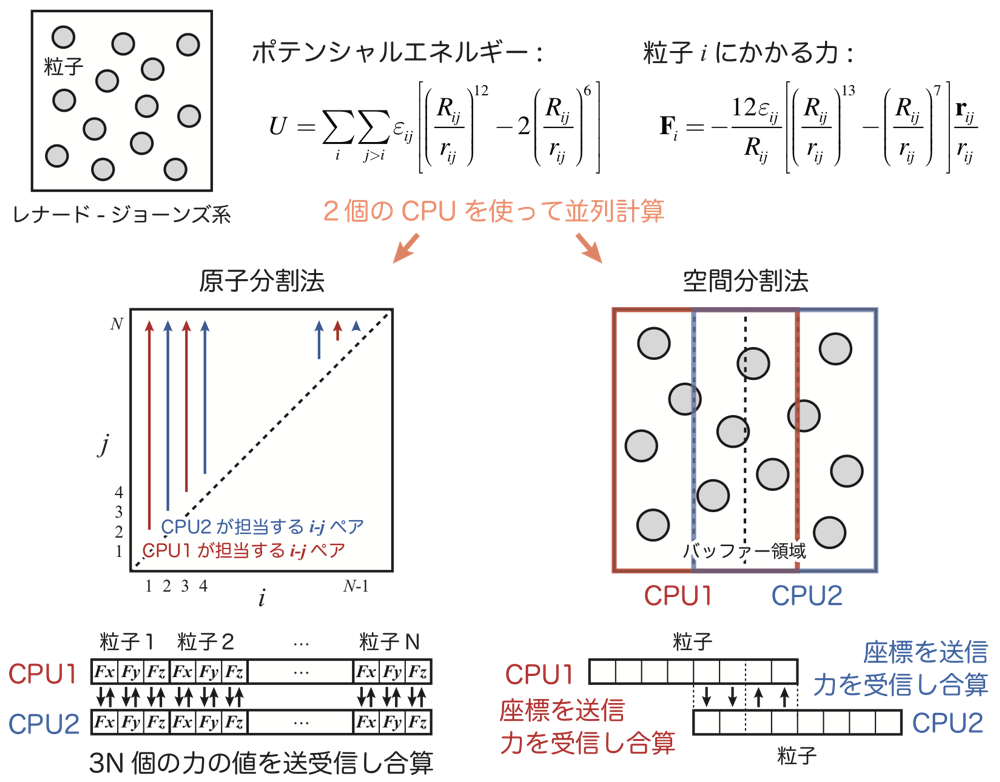
図2: 原子分割法と空間分割法による並列計算。原子分割法では、各 CPU は系に存在するすべての粒子の座標情報を保持しており、相互作用ペアリストを分割して並列計算を行う。各 CPU は各粒子にかかる力を計算した後、3N 個分の力の値を CPU 間で送受信し合算する。空間分割法では、各 CPU は自分のドメイン + バッファー領域内のみの粒子の座標情報を保持している。各 CPU はお互いにバッファー領域内の粒子の座標を送信し、力を受信して合算する。そのため、原子分割法と比べて大幅に通信量を減らすことができる。バッファー領域のサイズは、通常カットオフ距離またはそれ以上に設定する。
