

概要
代表的なセルピン病であるα1-アンチトリプシン欠乏症は、同蛋白質の変異体がポリマー化することで発症する遺伝病で、欧米では600–2500人に1人という極めて高い率で罹患する。2008年10月、本疾患の発症機構を解明し、且つこれまでのポリマー化研究を根こそぎ転覆させる驚くべき研究成果が発表された(Yamasaki et al., Nature)(1)。本稿では、α1-アンチトリプシンが属するセルピン・スーパーファミリーの構造と機能を概説し、Yamasaki らによる研究成果とセルピン病研究の動向について紹介する。
- セルピン病(serpinopathies)という病気をご存知でしょうか?
マイケル・ジャクソンやエルビス・プレスリーも苦しんだ恐ろしい病
(セルピン病をご存知の方はスキップして下さい) - Anfinsen のドグマに反する蛋白質・セルピンとは?
(セルピンの構造や機能についてご存知の方はスキップして下さい) - これまで信じられてきた、誰も疑わなかった旧ポリマー化メカニズム
- セルピン研究者を震撼させた全く新しいポリマー化メカニズム
- 解明されたミステリー治療へのラビリンス
1. セルピン病という病気をご存知でしょうか?
セルピン(serpins: serine protease inhibitor superfamily に属する蛋白質群)がポリマー化して起こる疾病群を総じて「セルピン病(serpinopathies)」と呼びます。ポリマー化したセルピンが本来の機能を果たさなくなって肺気腫や血栓症が起こったり、ポリマーが脳や肝臓に蓄積して認知症や肝硬変が起こったりします(表1a)。
最も有名なセルピン病であるα1-アンチトリプシン欠乏症(肺気腫、肝硬変)は、全世界で340万人以上の患者と1億人以上のキャリアーが存在すると言われています。マイケル・ジャクソンやエルビス・プレスリーなどの有名人の発病が度々話題となり、フレディック・ショパンは本疾患による肝硬変で亡くなったと言われています。ドラマ「ER」ではこの病気で呼吸困難になった急患が担ぎこまれる場面がありました。遺伝病ですが、その罹患率の高さから、大変身近で恐れられる疾患群の1つです — ただし、欧米では、の話です。
実はセルピン病は、極端に白色人種に偏って起こる疾患で、東洋人には稀です。中でも、島国・日本での発症数は極端に少なく、米国に20万人以上患者が存在するα1-アンチトリプシン欠乏症は、日本国内ではわずか数十人しか確認されていません。このような事情から、セルピン病、あるいはα1-アンチトリプシン欠乏症という言葉は、日本ではほとんど耳にすることはありません。
治療は、欠乏症に関しては正常型蛋白質(組換え型)の投与が標準法です。投与は生涯続きますが、遺伝子治療が難しい現状では、この手法しかありません。一方、ポリマーが生体内に蓄積して臓器不全を起こすタイプ(認知症、肝硬変)は、正常型蛋白質の投与では対処できません。肝硬変の場合は肝臓移植を行うと、正常型蛋白質が移植された肝臓から生合成され、治癒する場合があります。しかし認知症に関しては、残念ながら目下治療法はありません。
ちなみに、α1-アンチトリプシンの変異体はなんと80種以上も確認されています。それらの内、特に多いのは S 型(E264V, 南欧で28%)と Z 型(E342K, 北欧で4%)です。変異のタイプ、ホモかヘテロか、あるいは複数の変異体を有するかにより、発症の時期や症状の深刻さに差があり、煙草さえ吸わなければ生涯健康でいられるタイプもあります。また、変異体保有者は、たとえヘテロであっても肺がんに罹りやすいというデータがあります。よって、遺伝子チェックをすれば、病気の発症を防ぐことや、肺がん検診を頻繁に行うなどの対策がとれます。しかし、遺伝子診断の結果を、保険会社が利用することを禁じていない国があり(日本、米国など)、別の意味での不利益が生じる場合があります。
2. Anfinsen のドグマに反する蛋白質・セルピンとは?
セルピン・スーパーファミリーに属する蛋白質は、概ね、図1のような立体構造をしています。分子量4–6万、アミノ酸残基数は400前後のものが多く、ウイルスからヒトまで現在1000種以上確認されています。セリンプロテアーゼを阻害する蛋白質群(アンチトリプシン、アンチトロンビンなど)に共通する構造と言うことでこの名が付けられましたが、現在ではシステインプロテアーゼを阻害するものや、プロテアーゼ阻害機能を持たないもの(卵白アルブミンや HSP47)も見つかっています。
セルピンのプロテアーゼ阻害機構はいわゆる自殺基質方式で、非常にユニークな立体構造変化を伴うことから「マウストラップ」とも呼ばれています(図2)(2)。標的プロテアーゼの基質となるループ(反応中心ループ)をオトリのように差し出し、プロテアーゼがこれを切断すると、活性中心の Ser との間でアシル結合を形成、その後ループを挿入してプロテアーゼをトラップし、失活させます。
2つのβ-シートの間にループが挿入されて新たなβ-シートを形成するという劇的な構造変化は、ループ挿入型の方が Native 型よりもエネルギー的に安定であるために起こります。実際、ループ挿入型になると Tm 値は30℃以上上昇します。また、ループを切断しなくても、加温すれば Latent 型と呼ばれるループ挿入型に移行します(図3)。即ち、セルピンの Native 構造は、Anfinsen のドグマ1に反し、その一次配列にとっての最安定構造ではないのです。このような Native 型が準安定なフォールドをとる蛋白質は、セルピン以外にもありますが、自然界では極めて稀だと考えられています。そして、その稀な例が、プリオンやセルピンなどフォールディング病に関わる蛋白質に集中しています。
3. これまで信じられてきた、誰も疑わなかった旧ポリマー化メカニズム
セルピンの安定化に重要な事は、あのフラフラした反応中心ループが分子内部に納まることではありません。シャッター領域に適切なペプチドが挟まることが重要なのです。このことは、反応中心ループと同じ配列のペプチドを加えてインキュベーションしてやると、図3右のような複合体が形成され、安定化することから明らかです。
さて、病気の原因となるポリマー化はいかにして起こるのか? Lomas らは、セルピン病を引き起こす変異がシャッター領域付近に集中していることに注目し、図4a のモデルを発表しました(3,4)。病原性変異体ではシャッターが開きやすく、他の分子の反応中心ループが挿入され、この反応が連続してポリマーが伸長していくというものです。
その後、Lomas モデルを支持するデータが次々と発表されました。中間体 M* の結晶構造が発表され(PDB code: 1QMN)、ダミーのペプチドを外挿すれば(図3の複合体)ポリマー化しないことが明らかとなり、そしてついには、反応中心ループが切れてはいますが、ポリマーの結晶構造が発表されました(図4b)。1992年からの約16年間、このメカニズムを疑う者は誰もいませんでした。すべてのポリマー化に関わる研究はこのメカニズムを基に進められ、治療薬の開発も勿論、このメカニズムを前提としたものでした。
しかし、本当に誰も疑っていなかったのでしょうか? 筆者はセルピンをリフォールディングで以って研究する極めて少数派の人間なのですが(in vitro の主役は X 線結晶構造解析)、「セルピンの凝集体が蓄積する主因はミスフォールディングではないのか?」という疑惑を常々持っていました。病原性変異体のリフォールディング収率は野生型のそれに比べ極端に悪いのです(100分の1以下)。あるリフォールディング中間体まではほぼ同等なのですが、それ以降の構造回復が病原性変異体ではほとんど不可能に近い。しかしこの疑問は、セルピン・ポリマーの特性 —小胞体内に蓄積するにも関わらずアンフォールディング応答を引き起こさない— により一蹴され続けました。もしミスフォールディングが主因であれば、小胞体内の品質管理システムによる除去が起こるはずで、この点もまた「一度きちんとフォールドしたものがポリマー化する」という Lomas モデルの強い後ろ楯となりました。「私の実験がヘタクソだから収率が悪いのだろう。in vivo では、変異体だってきっと上手くフォールドできるに違いない」そう言い聞かせながら、Lomas ラボにて2008年の夏を向かえました。
4. セルピン研究者を震撼させた全く新しいポリマー化メカニズム
2008年8月、セルピン研究において歴史的な論文が Nature にアクセプトされました(1)。それはセルピン病の発症機作を鮮やかに解明する、Lomas モデルとは全く異なるポリマー化経路の発見でした。この論文の著者らは、Lomas グループと同じ建物の同じフロアーにラボを持ち、同じ遠心機と同じ化学天秤を共同で使う兄弟グループのような関係で、このプロジェクトに着手した当初は Lomas モデルになんら疑問を持っていませんでした。そもそも、この論文の最尾著者である Huntington 博士は、Lomas モデルを強く後押した図4b のポリマーの結晶構造を解いた張本人です。
これまで誰も疑わなかった Lomas 式ポリマーですが、その結晶構造(uncleaved 型)を解いた者はいませんでした。なぜなら、セルピンのポリマーはすぐに寄り集まって凝集体となるため、結晶化が困難だからです。しかし2004年、アンチトロンビン P80S 変異体がネックレスのように環状に繋がって自己完結するポリマーを作るという論文が発表され、これにヒントを得た Yamasaki 博士は、野生型アンチトロンビンから自己完結した安定なダイマーを調製し、見事そのX線結晶構造解析に成功したのです。
その構造は、全く誰も想像し得えなかったものでした(図5a)。シャッター領域において、4番と5番の2本のストランドがドメイン・スワッピングしていたのです。3番と5番の間が開き易いという証拠はこれまで散々見つかっていますが、5番のストランドが剥がれ易いという驚きの事実は、この結晶構造により初めて示されました。そして、ここからの仕事が素晴らしかった。この構造を見て、単に「セルピンはこんな構造も取り得る面白い蛋白質なんです」で終わらせなかった。この2本のストランド挿入は、このダイマーに限った事でもアンチトロンビンに限った事でもない、これこそが真のポリマー化のメカニズムであるとし、モデルを立て(図5b)、セルピン病の主役・α1-アンチトリプシンがこのメカニズムでポリマー化していることを見事に立証しました(図5c)。
2008年7月、Nature からのアクセプト通知に先立ち、この新しいポリマー化メカニズムは Gordon Research Conferences とセルピン専門学会(Serpins 2008)で口頭発表され、セルピン研究者達を震撼させました。なぜなら、この新しいメカニズムは、セルピン・ポリマーの性質を極めて良く反映し、Lomas モデルでは説明できなかった様々な矛盾を一気に解決してしまったからです(表2)。 Nature の News and Views では「Serpins’ mystery solved」とまで評されました(5)。一番の衝撃は、小胞体に蓄積するポリマーは、一旦フォールドされた Native 型から生じるのではなく、フォールディング中間体から形成されるという新説でした(図6)。病原性変異体のフォールディング効率は極端に悪いが、アンフォールディング応答は起こらない。蓄積型セルピン病の発症メカニズムが解明された瞬間でした。
5. 解明されたミステリー治療へのラビリンス
たった1つの論文が、たった1つの結晶構造が、これまで何百という論文により積上げられてきた理論を根本から覆す。サインエンスにおいて、しばしば起こる事なのかも知れません。ある日を境に交通法規が右側通行から左側通行に切り替わったように、研究者達の思考は一斉に Yamasaki モデルに流れました。Yamasaki モデルが発表される前にアクセプトされ、発表後に刊行となったセルピンの総説は、異例の修正再刊行となりました。当時、投稿準備中あるいは審査中の論文を抱えていた研究者達は、少なからずパニックになったかも知れません。
罹患者数最大のセルピン病であるα1-アンチトリプシン欠乏症には、巨大な患者団体が存在し、治療薬の開発には巨額の研究費が注ぎ込まれています。これまで、Lomas モデルを基にポリマー化を阻止する化合物の探索・創出が行われてきましたが(図4b)、Native コンフォメーションに基づく薬物設計や効果の検証は、少なくとも蓄積型(肝硬変)に関してはナンセンスなアプローチという烙印を押されてしまいました。今、多くのプロジェクトで大幅な見直しが迫られています。ポリマーの蓄積が認められない機能不全型(肺気腫)に関しては、Lomas/Yamasaki いずれのモデルが妥当か、あるいはそれ以外のポリマー化機構も関与するのか、議論は暫く続きそうですが、患者側からすれば、後者は正常型蛋白質の投与で対処できる訳で、より深刻な前者の治療法開発が何よりも急がれます。
Yamasaki モデルによりミステリーは解明されました。一方で、構造生物学的アプローチによる治療法の開発はほぼ振出しに戻りました。今後は遺伝子治療に重心が移るのか、あるいは近年発展目覚しい万能細胞にこそ道があるのか? 発症メカニズムが解明されてもなお、治療法確立までの道のりは遠い。これはフォールディング病に共通する悩ましい現実なのかも知れません。
参考文献
- Yamasaki, M. et al., Nature, 455, 1255–8 (2008)
- Huntington, J.A. et al., Nature, 407, 923–6 (2000)
- Lomas, D.A. et al., Nature, 357, 605–7 (1992)
- Davis, R.L. et al., Nature, 401, 376–9 (1999)
- Whisstock, J.C. & Bottomley, S.P., Nature, 455, 1189–90 (2008)
- Devlin, G.L. & Bottomley, S.P., Front Biosci., 10, 288–99 (2005)
-
Anfinsenのドグマ:蛋白質のフォールディング反応は自発的に起こり、Native 構造はエネルギー最小の状態である:by Christian B. Anfinsen(1972年ノーベル化学賞受賞) ↩
-
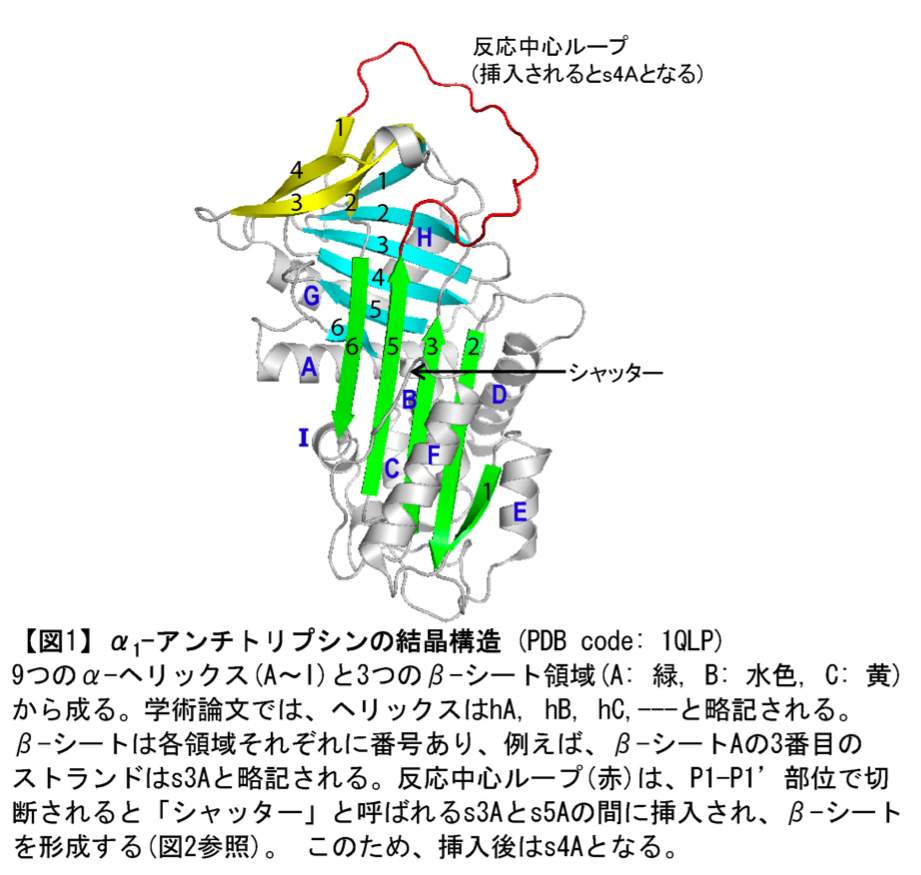
図1 -
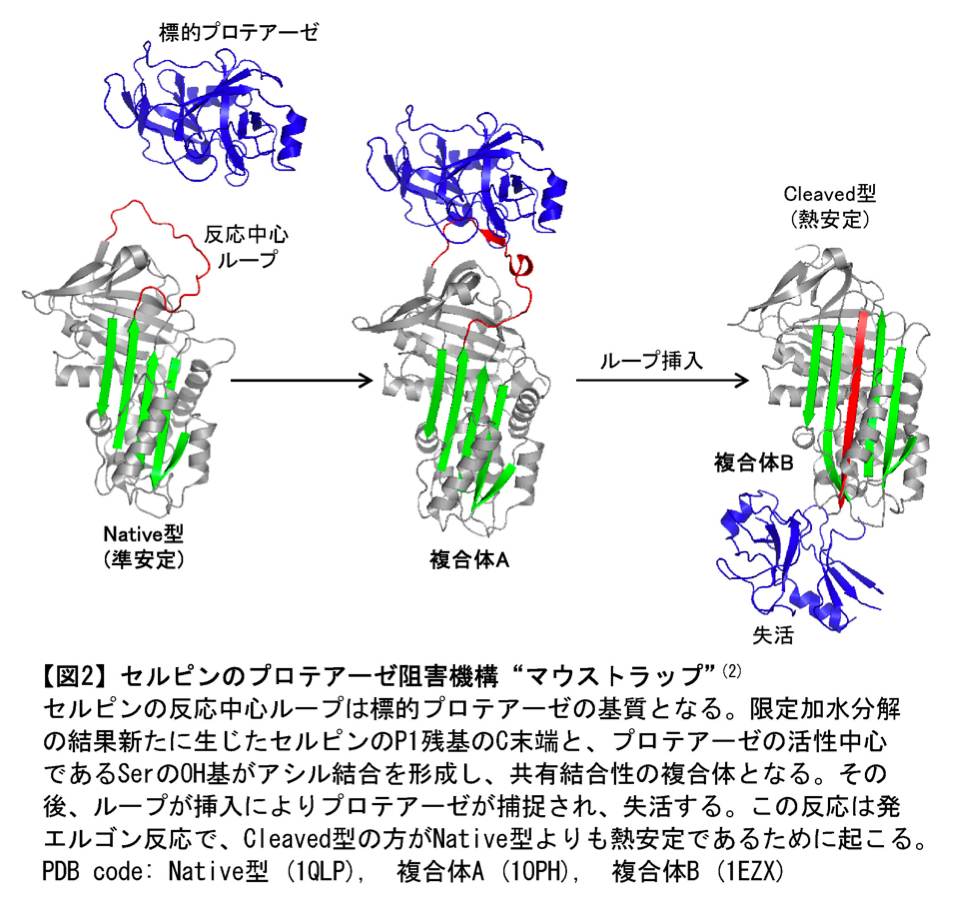
図2 -
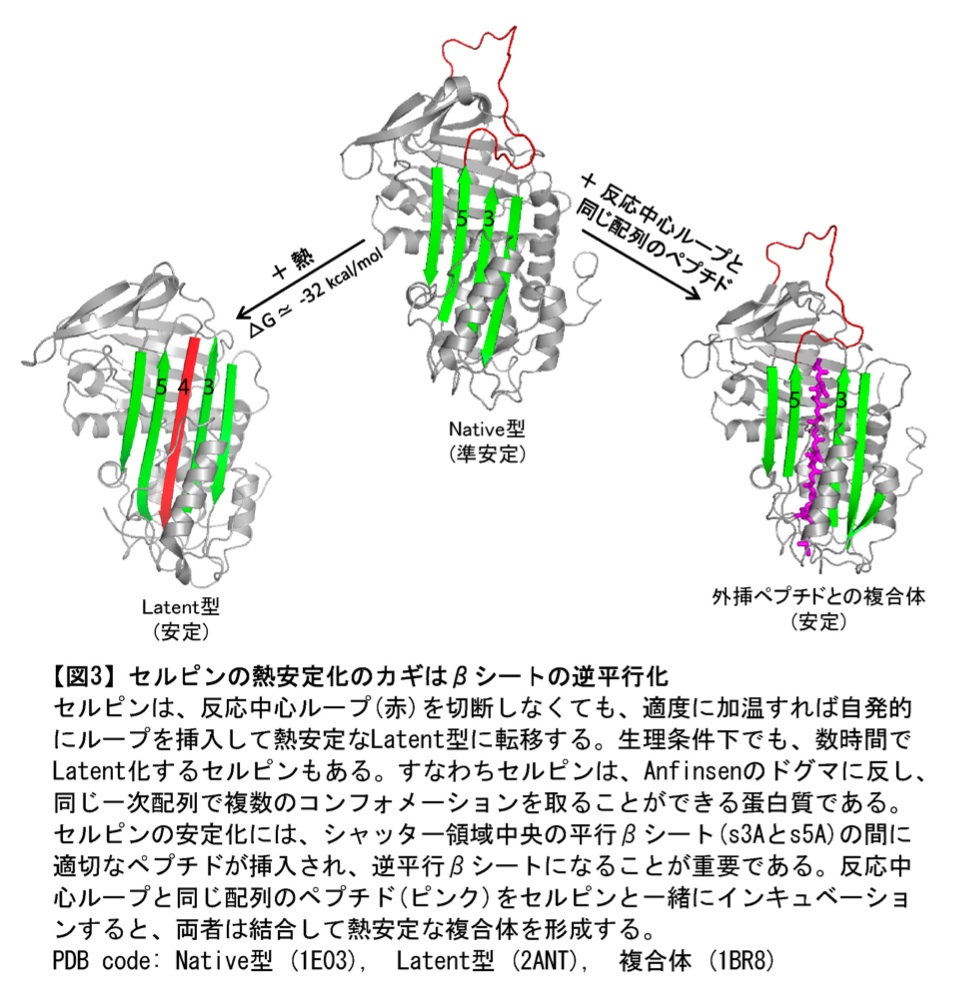
図3 -
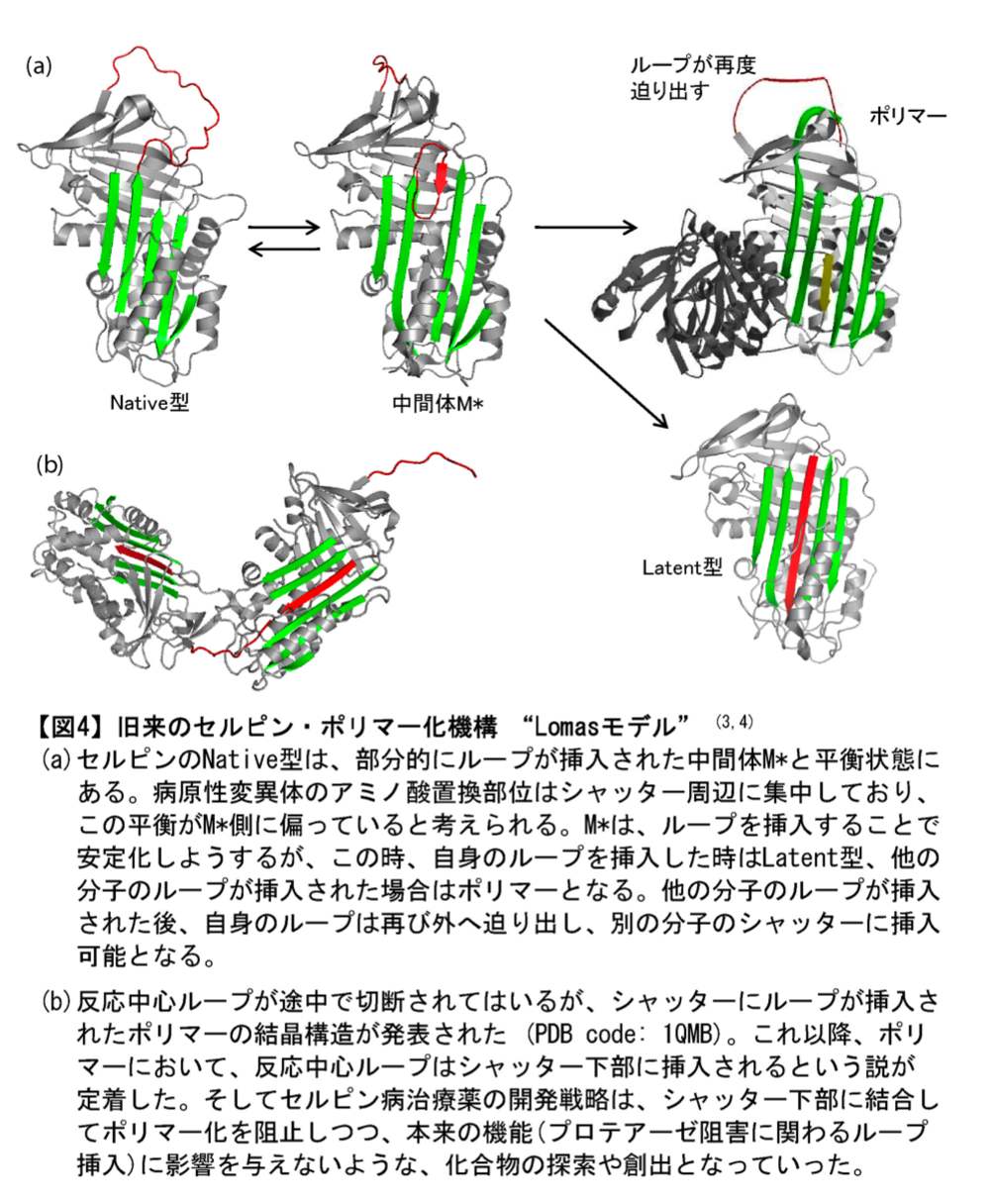
図4 -
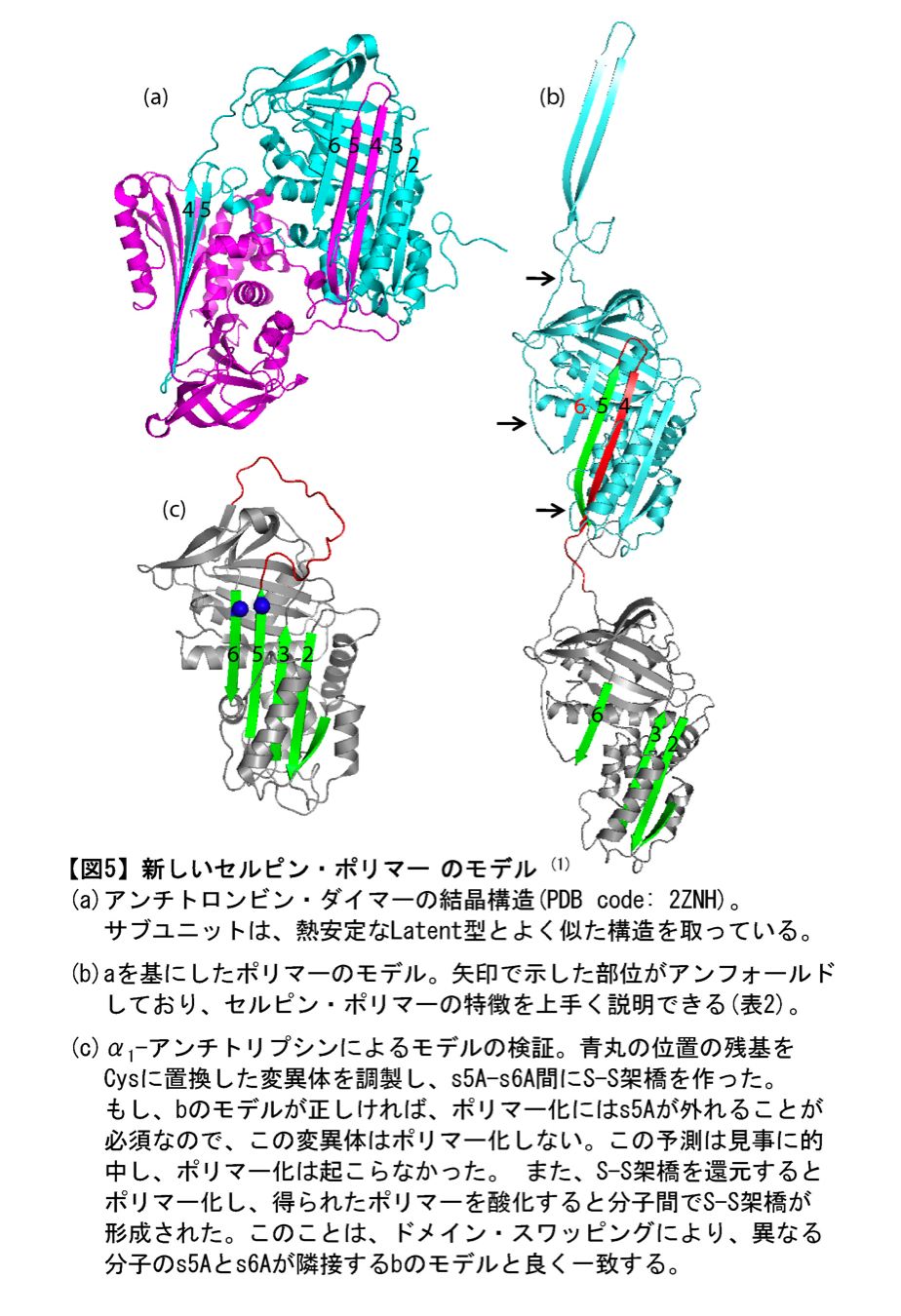
図5 -
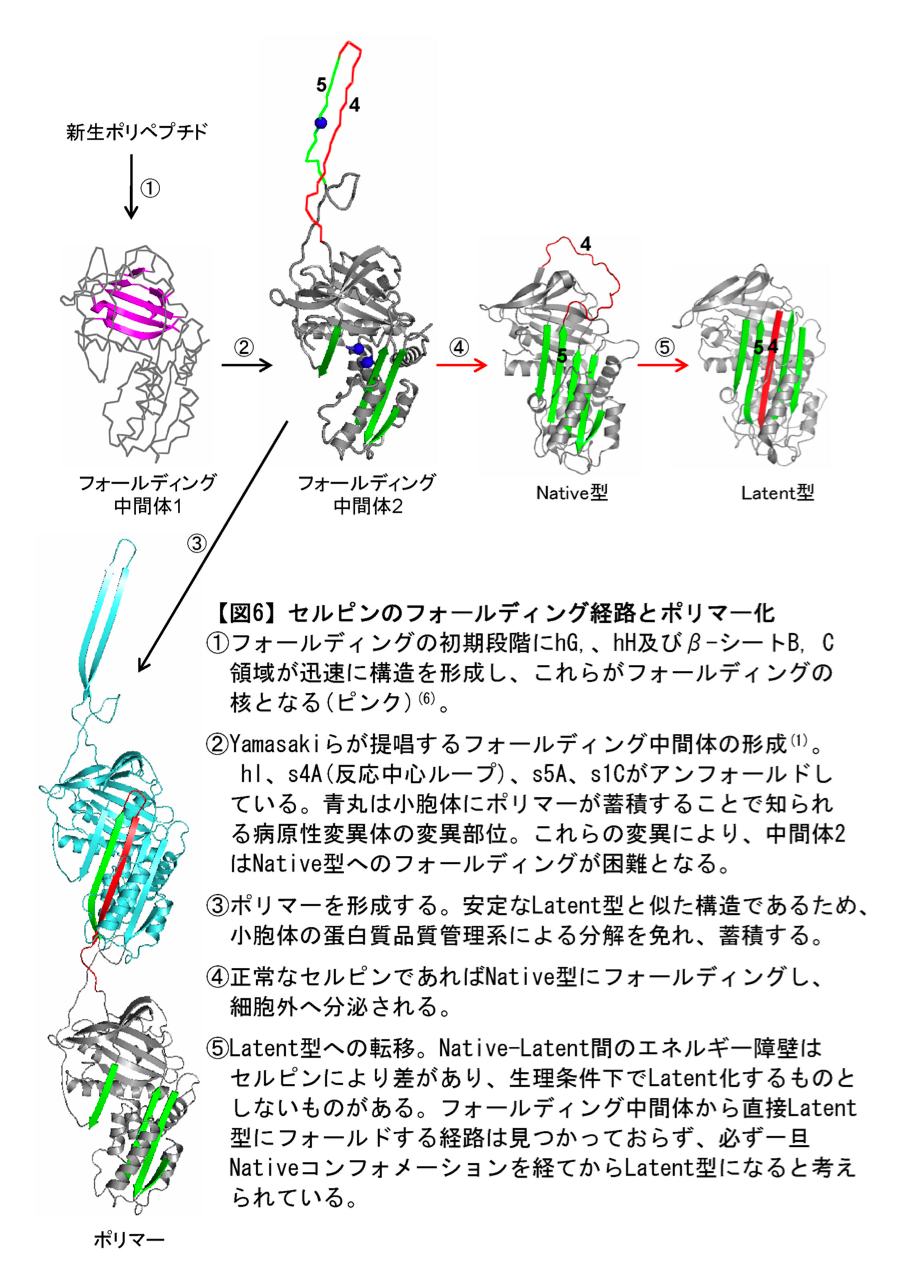
図6 -

表1 -
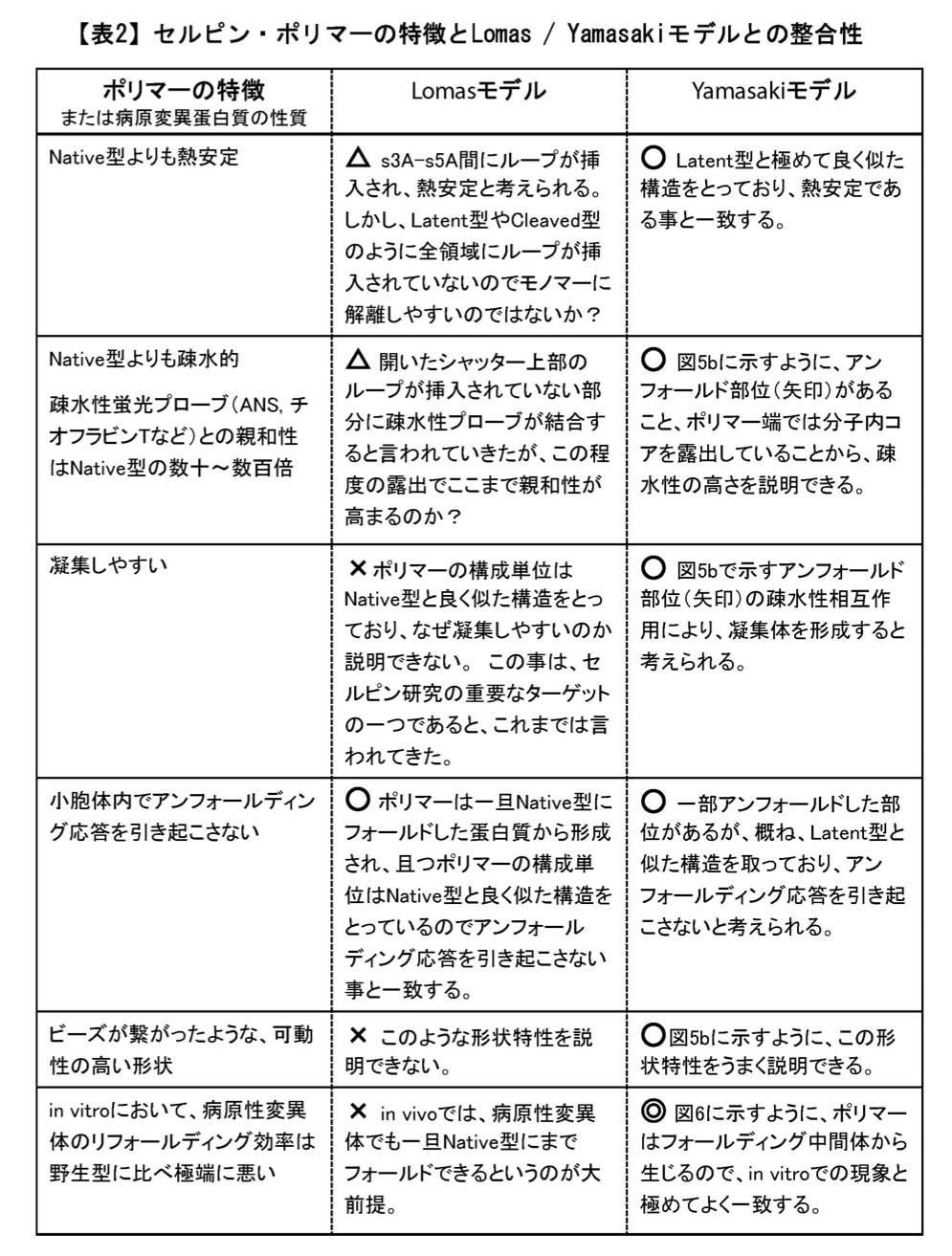
表2
概要
代表的なセルピン病であるα1-アンチトリプシン欠乏症は、同蛋白質の変異体がポリマー化することで発症する遺伝病で、欧米では600–2500人に1人という極めて高い率で罹患する。2008年10月、本疾患の発症機構を解明し、且つこれまでのポリマー化研究を根こそぎ転覆させる驚くべき研究成果が発表された(Yamasaki et al., Nature)(1)。本稿では、α1-アンチトリプシンが属するセルピン・スーパーファミリーの構造と機能を概説し、Yamasaki らによる研究成果とセルピン病研究の動向について紹介する。
- セルピン病(serpinopathies)という病気をご存知でしょうか?
マイケル・ジャクソンやエルビス・プレスリーも苦しんだ恐ろしい病
(セルピン病をご存知の方はスキップして下さい) - Anfinsen のドグマに反する蛋白質・セルピンとは?
(セルピンの構造や機能についてご存知の方はスキップして下さい) - これまで信じられてきた、誰も疑わなかった旧ポリマー化メカニズム
- セルピン研究者を震撼させた全く新しいポリマー化メカニズム
- 解明されたミステリー治療へのラビリンス
1. セルピン病という病気をご存知でしょうか?
セルピン(serpins: serine protease inhibitor superfamily に属する蛋白質群)がポリマー化して起こる疾病群を総じて「セルピン病(serpinopathies)」と呼びます。ポリマー化したセルピンが本来の機能を果たさなくなって肺気腫や血栓症が起こったり、ポリマーが脳や肝臓に蓄積して認知症や肝硬変が起こったりします(表1a)。
最も有名なセルピン病であるα1-アンチトリプシン欠乏症(肺気腫、肝硬変)は、全世界で340万人以上の患者と1億人以上のキャリアーが存在すると言われています。マイケル・ジャクソンやエルビス・プレスリーなどの有名人の発病が度々話題となり、フレディック・ショパンは本疾患による肝硬変で亡くなったと言われています。ドラマ「ER」ではこの病気で呼吸困難になった急患が担ぎこまれる場面がありました。遺伝病ですが、その罹患率の高さから、大変身近で恐れられる疾患群の1つです — ただし、欧米では、の話です。
実はセルピン病は、極端に白色人種に偏って起こる疾患で、東洋人には稀です。中でも、島国・日本での発症数は極端に少なく、米国に20万人以上患者が存在するα1-アンチトリプシン欠乏症は、日本国内ではわずか数十人しか確認されていません。このような事情から、セルピン病、あるいはα1-アンチトリプシン欠乏症という言葉は、日本ではほとんど耳にすることはありません。
治療は、欠乏症に関しては正常型蛋白質(組換え型)の投与が標準法です。投与は生涯続きますが、遺伝子治療が難しい現状では、この手法しかありません。一方、ポリマーが生体内に蓄積して臓器不全を起こすタイプ(認知症、肝硬変)は、正常型蛋白質の投与では対処できません。肝硬変の場合は肝臓移植を行うと、正常型蛋白質が移植された肝臓から生合成され、治癒する場合があります。しかし認知症に関しては、残念ながら目下治療法はありません。
ちなみに、α1-アンチトリプシンの変異体はなんと80種以上も確認されています。それらの内、特に多いのは S 型(E264V, 南欧で28%)と Z 型(E342K, 北欧で4%)です。変異のタイプ、ホモかヘテロか、あるいは複数の変異体を有するかにより、発症の時期や症状の深刻さに差があり、煙草さえ吸わなければ生涯健康でいられるタイプもあります。また、変異体保有者は、たとえヘテロであっても肺がんに罹りやすいというデータがあります。よって、遺伝子チェックをすれば、病気の発症を防ぐことや、肺がん検診を頻繁に行うなどの対策がとれます。しかし、遺伝子診断の結果を、保険会社が利用することを禁じていない国があり(日本、米国など)、別の意味での不利益が生じる場合があります。
2. Anfinsen のドグマに反する蛋白質・セルピンとは?
セルピン・スーパーファミリーに属する蛋白質は、概ね、図1のような立体構造をしています。分子量4–6万、アミノ酸残基数は400前後のものが多く、ウイルスからヒトまで現在1000種以上確認されています。セリンプロテアーゼを阻害する蛋白質群(アンチトリプシン、アンチトロンビンなど)に共通する構造と言うことでこの名が付けられましたが、現在ではシステインプロテアーゼを阻害するものや、プロテアーゼ阻害機能を持たないもの(卵白アルブミンや HSP47)も見つかっています。
セルピンのプロテアーゼ阻害機構はいわゆる自殺基質方式で、非常にユニークな立体構造変化を伴うことから「マウストラップ」とも呼ばれています(図2)(2)。標的プロテアーゼの基質となるループ(反応中心ループ)をオトリのように差し出し、プロテアーゼがこれを切断すると、活性中心の Ser との間でアシル結合を形成、その後ループを挿入してプロテアーゼをトラップし、失活させます。
2つのβ-シートの間にループが挿入されて新たなβ-シートを形成するという劇的な構造変化は、ループ挿入型の方が Native 型よりもエネルギー的に安定であるために起こります。実際、ループ挿入型になると Tm 値は30℃以上上昇します。また、ループを切断しなくても、加温すれば Latent 型と呼ばれるループ挿入型に移行します(図3)。即ち、セルピンの Native 構造は、Anfinsen のドグマ1に反し、その一次配列にとっての最安定構造ではないのです。このような Native 型が準安定なフォールドをとる蛋白質は、セルピン以外にもありますが、自然界では極めて稀だと考えられています。そして、その稀な例が、プリオンやセルピンなどフォールディング病に関わる蛋白質に集中しています。
3. これまで信じられてきた、誰も疑わなかった旧ポリマー化メカニズム
セルピンの安定化に重要な事は、あのフラフラした反応中心ループが分子内部に納まることではありません。シャッター領域に適切なペプチドが挟まることが重要なのです。このことは、反応中心ループと同じ配列のペプチドを加えてインキュベーションしてやると、図3右のような複合体が形成され、安定化することから明らかです。
さて、病気の原因となるポリマー化はいかにして起こるのか? Lomas らは、セルピン病を引き起こす変異がシャッター領域付近に集中していることに注目し、図4a のモデルを発表しました(3,4)。病原性変異体ではシャッターが開きやすく、他の分子の反応中心ループが挿入され、この反応が連続してポリマーが伸長していくというものです。
その後、Lomas モデルを支持するデータが次々と発表されました。中間体 M* の結晶構造が発表され(PDB code: 1QMN)、ダミーのペプチドを外挿すれば(図3の複合体)ポリマー化しないことが明らかとなり、そしてついには、反応中心ループが切れてはいますが、ポリマーの結晶構造が発表されました(図4b)。1992年からの約16年間、このメカニズムを疑う者は誰もいませんでした。すべてのポリマー化に関わる研究はこのメカニズムを基に進められ、治療薬の開発も勿論、このメカニズムを前提としたものでした。
しかし、本当に誰も疑っていなかったのでしょうか? 筆者はセルピンをリフォールディングで以って研究する極めて少数派の人間なのですが(in vitro の主役は X 線結晶構造解析)、「セルピンの凝集体が蓄積する主因はミスフォールディングではないのか?」という疑惑を常々持っていました。病原性変異体のリフォールディング収率は野生型のそれに比べ極端に悪いのです(100分の1以下)。あるリフォールディング中間体まではほぼ同等なのですが、それ以降の構造回復が病原性変異体ではほとんど不可能に近い。しかしこの疑問は、セルピン・ポリマーの特性 —小胞体内に蓄積するにも関わらずアンフォールディング応答を引き起こさない— により一蹴され続けました。もしミスフォールディングが主因であれば、小胞体内の品質管理システムによる除去が起こるはずで、この点もまた「一度きちんとフォールドしたものがポリマー化する」という Lomas モデルの強い後ろ楯となりました。「私の実験がヘタクソだから収率が悪いのだろう。in vivo では、変異体だってきっと上手くフォールドできるに違いない」そう言い聞かせながら、Lomas ラボにて2008年の夏を向かえました。
4. セルピン研究者を震撼させた全く新しいポリマー化メカニズム
2008年8月、セルピン研究において歴史的な論文が Nature にアクセプトされました(1)。それはセルピン病の発症機作を鮮やかに解明する、Lomas モデルとは全く異なるポリマー化経路の発見でした。この論文の著者らは、Lomas グループと同じ建物の同じフロアーにラボを持ち、同じ遠心機と同じ化学天秤を共同で使う兄弟グループのような関係で、このプロジェクトに着手した当初は Lomas モデルになんら疑問を持っていませんでした。そもそも、この論文の最尾著者である Huntington 博士は、Lomas モデルを強く後押した図4b のポリマーの結晶構造を解いた張本人です。
これまで誰も疑わなかった Lomas 式ポリマーですが、その結晶構造(uncleaved 型)を解いた者はいませんでした。なぜなら、セルピンのポリマーはすぐに寄り集まって凝集体となるため、結晶化が困難だからです。しかし2004年、アンチトロンビン P80S 変異体がネックレスのように環状に繋がって自己完結するポリマーを作るという論文が発表され、これにヒントを得た Yamasaki 博士は、野生型アンチトロンビンから自己完結した安定なダイマーを調製し、見事そのX線結晶構造解析に成功したのです。
その構造は、全く誰も想像し得えなかったものでした(図5a)。シャッター領域において、4番と5番の2本のストランドがドメイン・スワッピングしていたのです。3番と5番の間が開き易いという証拠はこれまで散々見つかっていますが、5番のストランドが剥がれ易いという驚きの事実は、この結晶構造により初めて示されました。そして、ここからの仕事が素晴らしかった。この構造を見て、単に「セルピンはこんな構造も取り得る面白い蛋白質なんです」で終わらせなかった。この2本のストランド挿入は、このダイマーに限った事でもアンチトロンビンに限った事でもない、これこそが真のポリマー化のメカニズムであるとし、モデルを立て(図5b)、セルピン病の主役・α1-アンチトリプシンがこのメカニズムでポリマー化していることを見事に立証しました(図5c)。
2008年7月、Nature からのアクセプト通知に先立ち、この新しいポリマー化メカニズムは Gordon Research Conferences とセルピン専門学会(Serpins 2008)で口頭発表され、セルピン研究者達を震撼させました。なぜなら、この新しいメカニズムは、セルピン・ポリマーの性質を極めて良く反映し、Lomas モデルでは説明できなかった様々な矛盾を一気に解決してしまったからです(表2)。 Nature の News and Views では「Serpins’ mystery solved」とまで評されました(5)。一番の衝撃は、小胞体に蓄積するポリマーは、一旦フォールドされた Native 型から生じるのではなく、フォールディング中間体から形成されるという新説でした(図6)。病原性変異体のフォールディング効率は極端に悪いが、アンフォールディング応答は起こらない。蓄積型セルピン病の発症メカニズムが解明された瞬間でした。
5. 解明されたミステリー治療へのラビリンス
たった1つの論文が、たった1つの結晶構造が、これまで何百という論文により積上げられてきた理論を根本から覆す。サインエンスにおいて、しばしば起こる事なのかも知れません。ある日を境に交通法規が右側通行から左側通行に切り替わったように、研究者達の思考は一斉に Yamasaki モデルに流れました。Yamasaki モデルが発表される前にアクセプトされ、発表後に刊行となったセルピンの総説は、異例の修正再刊行となりました。当時、投稿準備中あるいは審査中の論文を抱えていた研究者達は、少なからずパニックになったかも知れません。
罹患者数最大のセルピン病であるα1-アンチトリプシン欠乏症には、巨大な患者団体が存在し、治療薬の開発には巨額の研究費が注ぎ込まれています。これまで、Lomas モデルを基にポリマー化を阻止する化合物の探索・創出が行われてきましたが(図4b)、Native コンフォメーションに基づく薬物設計や効果の検証は、少なくとも蓄積型(肝硬変)に関してはナンセンスなアプローチという烙印を押されてしまいました。今、多くのプロジェクトで大幅な見直しが迫られています。ポリマーの蓄積が認められない機能不全型(肺気腫)に関しては、Lomas/Yamasaki いずれのモデルが妥当か、あるいはそれ以外のポリマー化機構も関与するのか、議論は暫く続きそうですが、患者側からすれば、後者は正常型蛋白質の投与で対処できる訳で、より深刻な前者の治療法開発が何よりも急がれます。
Yamasaki モデルによりミステリーは解明されました。一方で、構造生物学的アプローチによる治療法の開発はほぼ振出しに戻りました。今後は遺伝子治療に重心が移るのか、あるいは近年発展目覚しい万能細胞にこそ道があるのか? 発症メカニズムが解明されてもなお、治療法確立までの道のりは遠い。これはフォールディング病に共通する悩ましい現実なのかも知れません。
参考文献
- Yamasaki, M. et al., Nature, 455, 1255–8 (2008)
- Huntington, J.A. et al., Nature, 407, 923–6 (2000)
- Lomas, D.A. et al., Nature, 357, 605–7 (1992)
- Davis, R.L. et al., Nature, 401, 376–9 (1999)
- Whisstock, J.C. & Bottomley, S.P., Nature, 455, 1189–90 (2008)
- Devlin, G.L. & Bottomley, S.P., Front Biosci., 10, 288–99 (2005)
-
Anfinsenのドグマ:蛋白質のフォールディング反応は自発的に起こり、Native 構造はエネルギー最小の状態である:by Christian B. Anfinsen(1972年ノーベル化学賞受賞) ↩
